

Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution
- PMID: 17568002
- PMCID: PMC1891343
- DOI: 10.1101/gr.5586307
Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution
Abstract
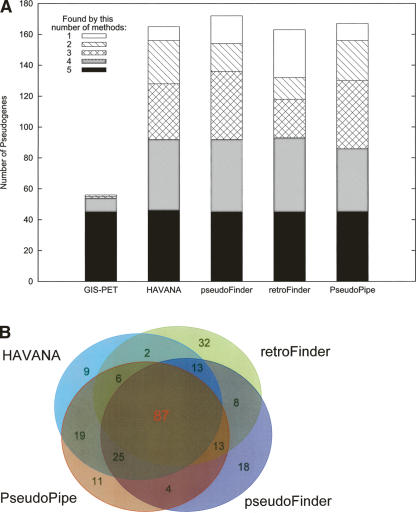
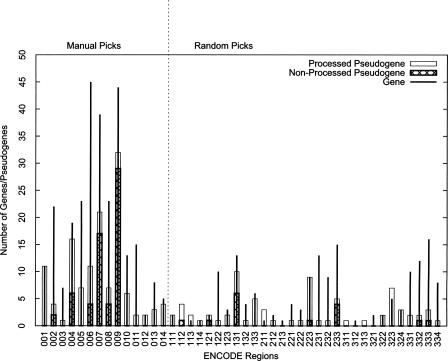
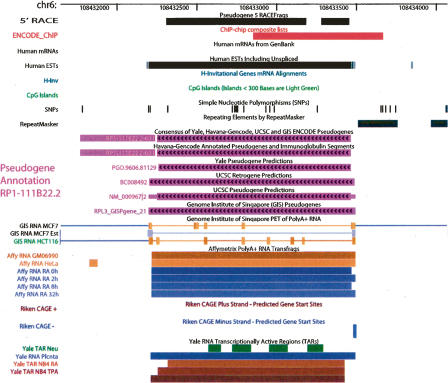
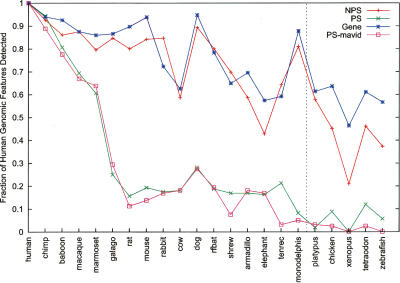
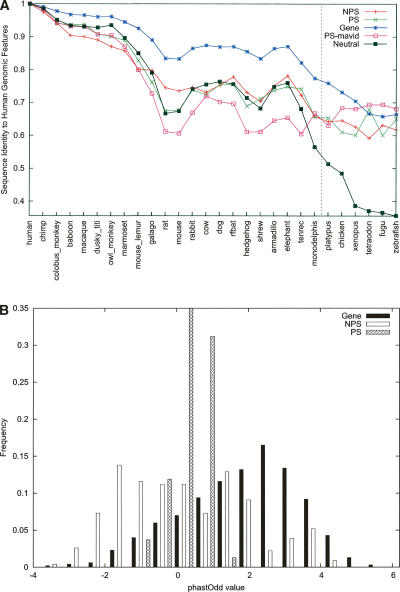
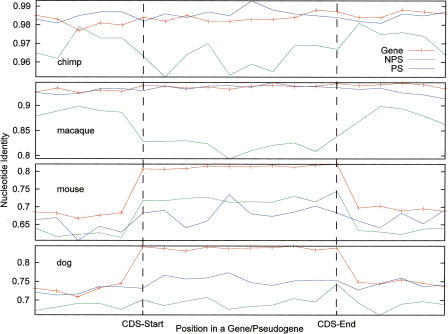
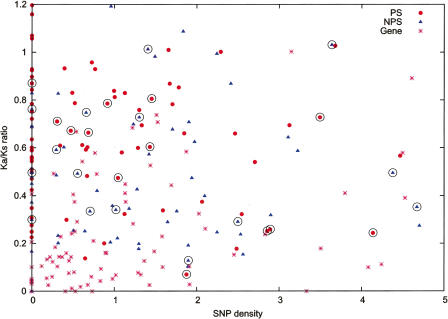
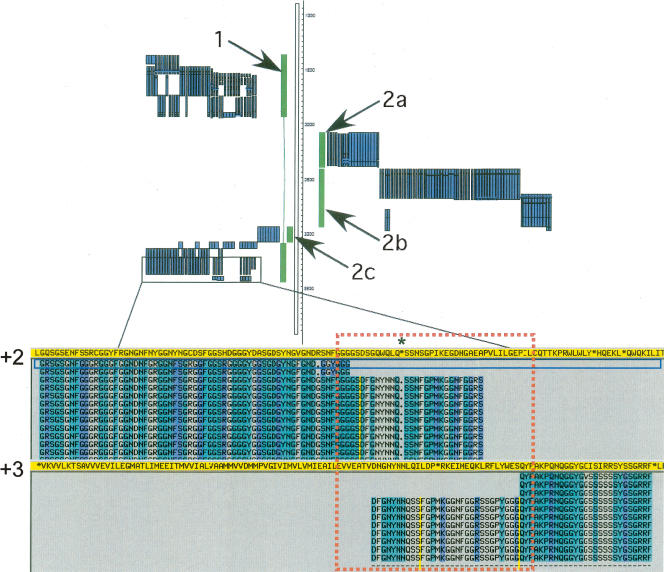
Arising from either retrotransposition or genomic duplication of functional genes, pseudogenes are "genomic fossils" valuable for exploring the dynamics and evolution of genes and genomes. Pseudogene identification is an important problem in computational genomics, and is also critical for obtaining an accurate picture of a genome's structure and function. However, no consensus computational scheme for defining and detecting pseudogenes has been developed thus far. As part of the ENCyclopedia Of DNA Elements (ENCODE) project, we have compared several distinct pseudogene annotation strategies and found that different approaches and parameters often resulted in rather distinct sets of pseudogenes. We subsequently developed a consensus approach for annotating pseudogenes (derived from protein coding genes) in the ENCODE regions, resulting in 201 pseudogenes, two-thirds of which originated from retrotransposition. A survey of orthologs for these pseudogenes in 28 vertebrate genomes showed that a significant fraction ( approximately 80%) of the processed pseudogenes are primate-specific sequences, highlighting the increasing retrotransposition activity in primates. Analysis of sequence conservation and variation also demonstrated that most pseudogenes evolve neutrally, and processed pseudogenes appear to have lost their coding potential immediately or soon after their emergence. In order to explore the functional implication of pseudogene prevalence, we have extensively examined the transcriptional activity of the ENCODE pseudogenes. We performed systematic series of pseudogene-specific RACE analyses. These, together with complementary evidence derived from tiling microarrays and high throughput sequencing, demonstrated that at least a fifth of the 201 pseudogenes are transcribed in one or more cell lines or tissues.
Figures

References
-
- Bairoch A., Apweiler R., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Apweiler R., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Gasteiger E., Huang H., Lopez R., Magrane M., Huang H., Lopez R., Magrane M., Lopez R., Magrane M., Magrane M., et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–D159. - PMC - PubMed
-
- Balakirev E.S., Ayala F.J., Ayala F.J. Pseudogenes: Are they “junk” or functional DNA? Annu. Rev. Genet. 2003;37:123–151. - PubMed
-
- Bertone P., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Tongprasit W., Samanta M., Weissman S., Samanta M., Weissman S., Weissman S., et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. - PubMed
-
- Bischof J.M., Chiang A.P., Scheetz T.E., Stone E.M., Casavant T.L., Sheffield V.C., Braun T.A., Chiang A.P., Scheetz T.E., Stone E.M., Casavant T.L., Sheffield V.C., Braun T.A., Scheetz T.E., Stone E.M., Casavant T.L., Sheffield V.C., Braun T.A., Stone E.M., Casavant T.L., Sheffield V.C., Braun T.A., Casavant T.L., Sheffield V.C., Braun T.A., Sheffield V.C., Braun T.A., Braun T.A. Genome-wide identification of pseudogenes capable of disease-causing gene conversion. Hum. Mutat. 2006;27:545–552. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials