

Mapping the chromosomal targets of STAT1 by Sequence Tag Analysis of Genomic Enrichment (STAGE)
- PMID: 17568006
- PMCID: PMC1891349
- DOI: 10.1101/gr.5574907
Mapping the chromosomal targets of STAT1 by Sequence Tag Analysis of Genomic Enrichment (STAGE)
Abstract
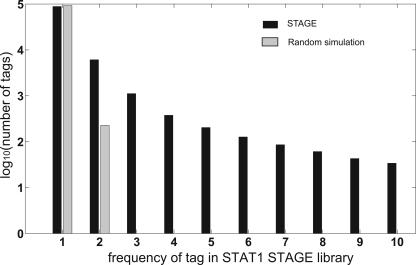
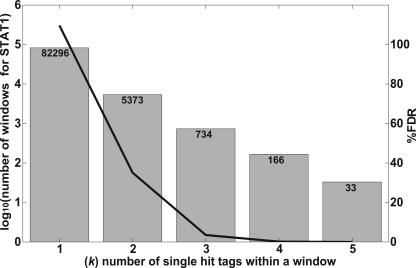
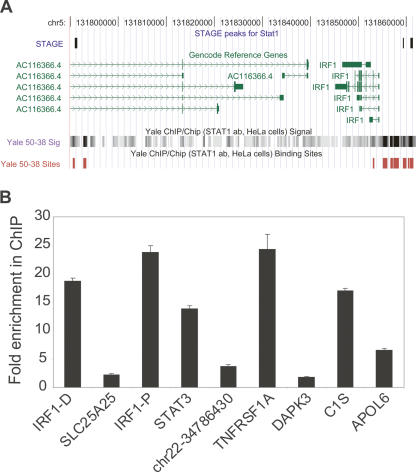
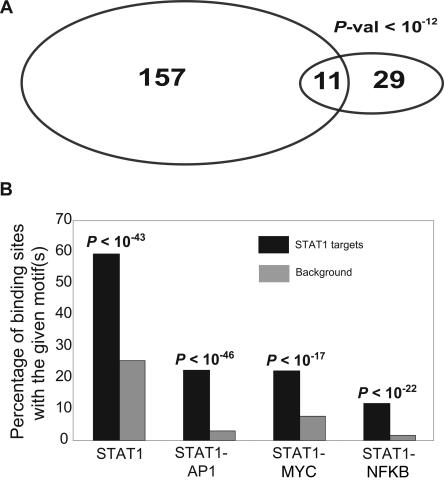
Identifying the genome-wide binding sites of transcription factors is important in deciphering transcriptional regulatory networks. ChIP-chip (Chromatin immunoprecipitation combined with microarrays) has been widely used to map transcription factor binding sites in the human genome. However, whole genome ChIP-chip analysis is still technically challenging in vertebrates. We recently developed STAGE as an unbiased method for identifying transcription factor binding sites in the genome. STAGE is conceptually based on SAGE, except that the input is ChIP-enriched DNA. In this study, we implemented an improved sequencing strategy and analysis methods and applied STAGE to map the genomic binding profile of the transcription factor STAT1 after interferon treatment. STAT1 is mainly responsible for mediating the cellular responses to interferons, such as cell proliferation, apoptosis, immune surveillance, and immune responses. We present novel algorithms for STAGE tag analysis to identify enriched loci with high specificity, as verified by quantitative ChIP. STAGE identified several previously unknown STAT1 target genes, many of which are involved in mediating the response to interferon-gamma signaling. STAGE is thus a viable method for identifying the chromosomal targets of transcription factors and generating meaningful biological hypotheses that further our understanding of transcriptional regulatory networks.
Figures
References
-
- Adhikary S., Eilers M., Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005;6:635–645. - PubMed
-
- Burch L.R., Scott M., Pohler E., Meek D., Hupp T., Scott M., Pohler E., Meek D., Hupp T., Pohler E., Meek D., Hupp T., Meek D., Hupp T., Hupp T. Phage-peptide display identifies the interferon-responsive, death-activated protein kinase family as a novel modifier of MDM2 and p21WAF1. J. Mol. Biol. 2004;337:115–128. - PubMed
-
- Cawley S., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Sementchenko V., Cheng J., Williams A.J., Cheng J., Williams A.J., Williams A.J., et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. - PubMed
-
- Cormen T., Leiserson C., Rivest R., Leiserson C., Rivest R., Rivest R. Introduction to algorithms. MIT Press; Cambridge, MA: 2001.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous