

A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites
- PMID: 17570152
- PMCID: PMC1892088
- DOI: 10.1186/1471-2105-8-S4-S9
A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites
Abstract
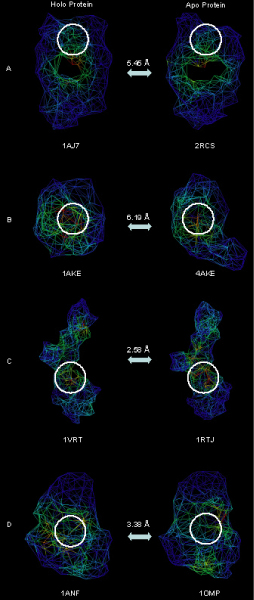
Background: An accurate description of protein shape derived from protein structure is necessary to establish an understanding of protein-ligand interactions, which in turn will lead to improved methods for protein-ligand docking and binding site analysis. Most current shape descriptors characterize only the local properties of protein structure using an all-atom representation and are slow to compute. We need new shape descriptors that have the ability to capture both local and global structural information, are robust for application to models and low quality structures and are computationally efficient to permit high throughput analysis of protein structures.
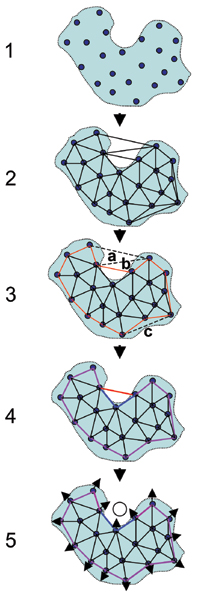
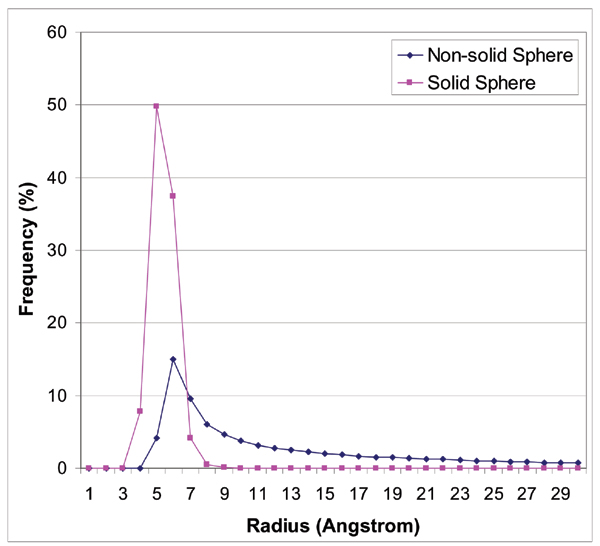
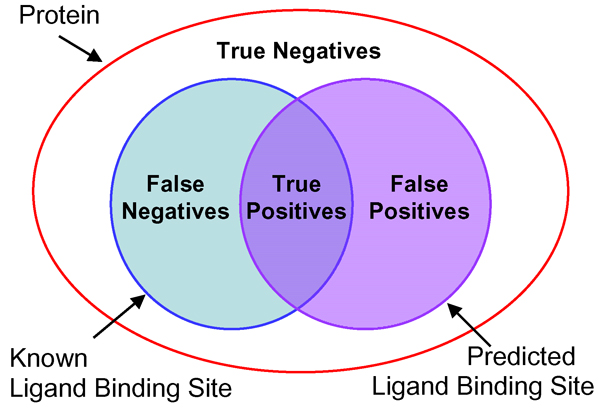
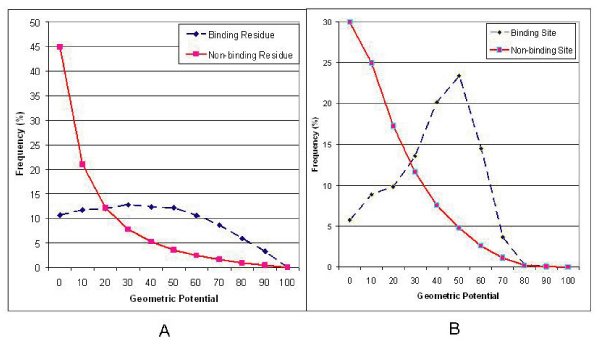
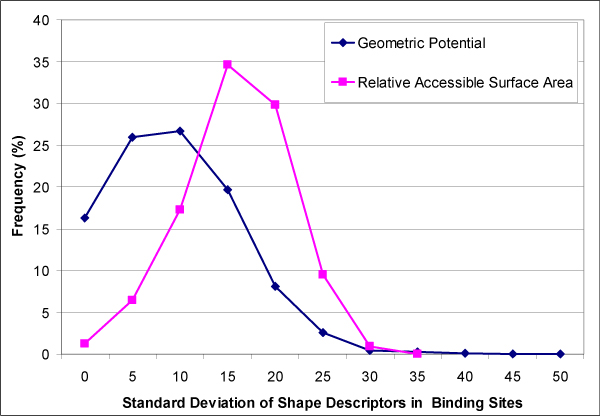
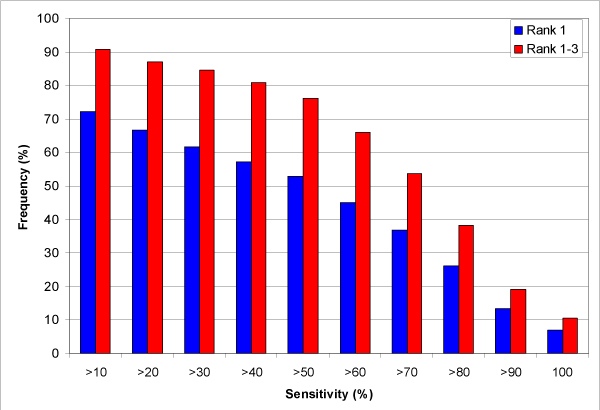
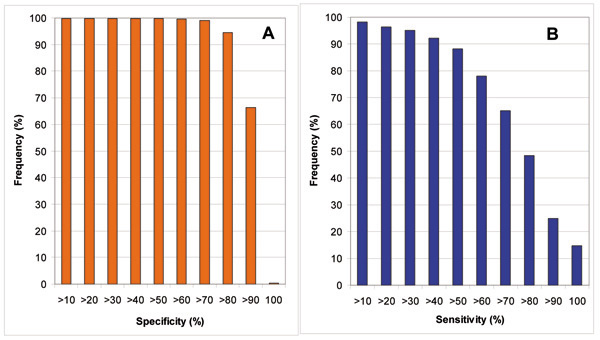
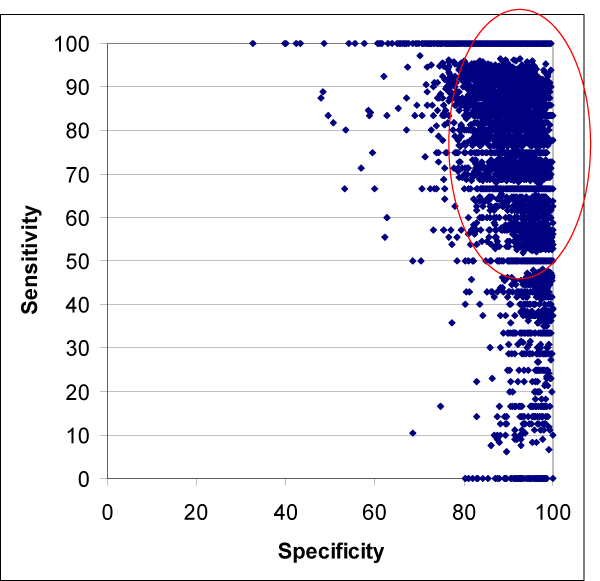
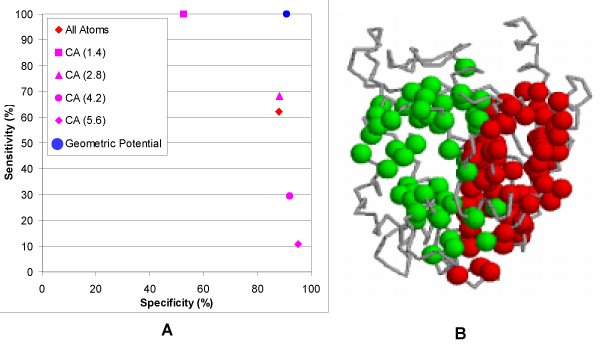
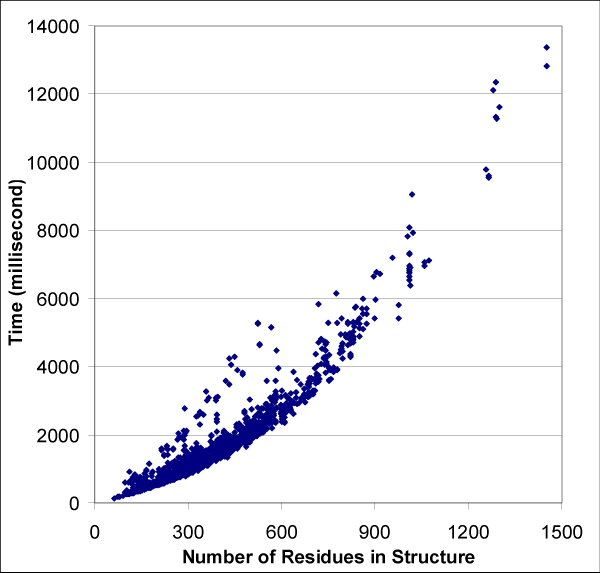
Results: We introduce a new shape description that requires only the Calpha atoms to represent the protein structure, thus making it both fast and suitable for use on models and low quality structures. The notion of a geometric potential is introduced to quantitatively describe the shape of the structure. This geometric potential is dependent on both the global shape of the protein structure as well as the surrounding environment of each residue. When applying the geometric potential for binding site prediction, approximately 85% of known binding sites can be accurately identified with above 50% residue coverage and 80% specificity. Moreover, the algorithm is fast enough for proteome-scale applications. Proteins with fewer than 500 amino acids can be scanned in less than two seconds.
Conclusion: The reduced representation of the protein structure combined with the geometric potential provides a fast, quantitative description of protein-ligand binding sites with potential for use in large-scale predictions, comparisons and analysis.
Figures
References
-
- Agarwal PK, Edelsbrunner H, Harer J, Wang Y. Extreme elevation on a 2-manifold. Symp Comp Geo. 2004;20:357–365.
-
- Hendrix DK, Kuntz ID. Surface solid angle-based site points for molecular docking. Pac Symp Biocomput: 1998. 1998. pp. 317–326. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
