

Accurate prediction of protein secondary structure and solvent accessibility by consensus combiners of sequence and structure information
- PMID: 17570843
- PMCID: PMC1913928
- DOI: 10.1186/1471-2105-8-201
Accurate prediction of protein secondary structure and solvent accessibility by consensus combiners of sequence and structure information
Abstract
Background: Structural properties of proteins such as secondary structure and solvent accessibility contribute to three-dimensional structure prediction, not only in the ab initio case but also when homology information to known structures is available. Structural properties are also routinely used in protein analysis even when homology is available, largely because homology modelling is lower throughput than, say, secondary structure prediction. Nonetheless, predictors of secondary structure and solvent accessibility are virtually always ab initio.
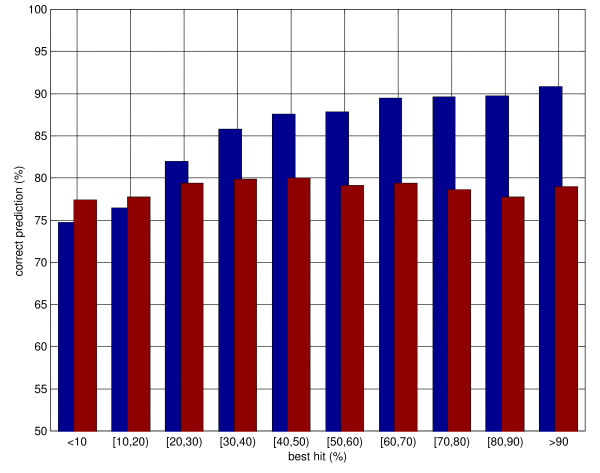
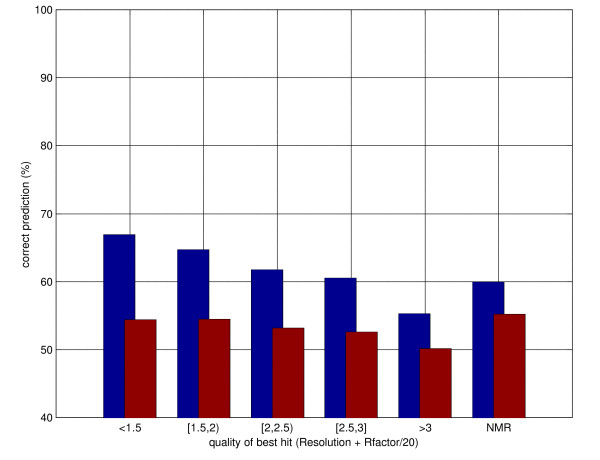
Results: Here we develop high-throughput machine learning systems for the prediction of protein secondary structure and solvent accessibility that exploit homology to proteins of known structure, where available, in the form of simple structural frequency profiles extracted from sets of PDB templates. We compare these systems to their state-of-the-art ab initio counterparts, and with a number of baselines in which secondary structures and solvent accessibilities are extracted directly from the templates. We show that structural information from templates greatly improves secondary structure and solvent accessibility prediction quality, and that, on average, the systems significantly enrich the information contained in the templates. For sequence similarity exceeding 30%, secondary structure prediction quality is approximately 90%, close to its theoretical maximum, and 2-class solvent accessibility roughly 85%. Gains are robust with respect to template selection noise, and significant for marginal sequence similarity and for short alignments, supporting the claim that these improved predictions may prove beneficial beyond the case in which clear homology is available.
Conclusion: The predictive system are publicly available at the address http://distill.ucd.ie.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
