

Automatic extraction of reliable regions from multiple sequence alignments
- PMID: 17570868
- PMCID: PMC1892097
- DOI: 10.1186/1471-2105-8-S5-S9
Automatic extraction of reliable regions from multiple sequence alignments
Abstract
Background: High quality multiple alignments are crucial in the transfer of annotation from one genome to another. Multiple alignment methods strive to achieve ever increasing levels of average accuracy on benchmark sets while the accuracy of individual alignments is often overlooked.
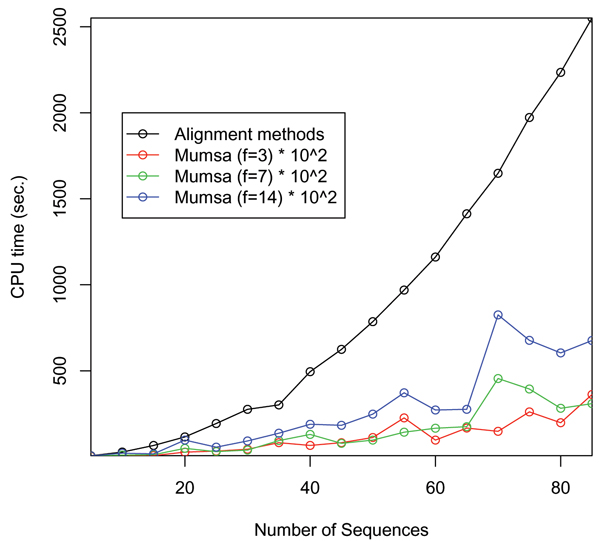
Results: We have previously developed a method to automatically assess the accuracy and overall difficulty of multiple alignments. This was achieved by a per-residue comparison between alternate alignments of the same sequences. Here we present a key extension to this method, an algorithm to extract similarly aligned regions from several alignments and merge them into a new consensus alignment.
Conclusion: We demonstrate that the fraction of correctly aligned residues within the resulting alignments is increased by 25-100 percent compared to the original input alignments, as only the most reliably aligned parts are considered.
Figures
References
-
- Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. http://www.genome.org/cgi/content/abstract/15/2/330 - DOI - PMC - PubMed
-
- Katoh K, Kuma Ki, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucl Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. http://nar.oxfordjournals.org/cgi/content/abstract/33/2/511 - DOI - PMC - PubMed
-
- Lassmann T, Sonnhammer E. Kalign – an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 2005;6:298. doi: 10.1186/1471-2105-6-298. http://www.biomedcentral.com/1471-2105/6/298 - DOI - PMC - PubMed
-
- Wallace IM, O'Sullivan O, Higgins DG, Notredame C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucl Acids Res. 2006;34:1692–1699. doi: 10.1093/nar/gkl091. http://nar.oxfordjournals.org/cgi/content/abstract/34/6/1692 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
