

Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project
- PMID: 17571346
- PMCID: PMC2212820
- DOI: 10.1038/nature05874
Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project
Abstract
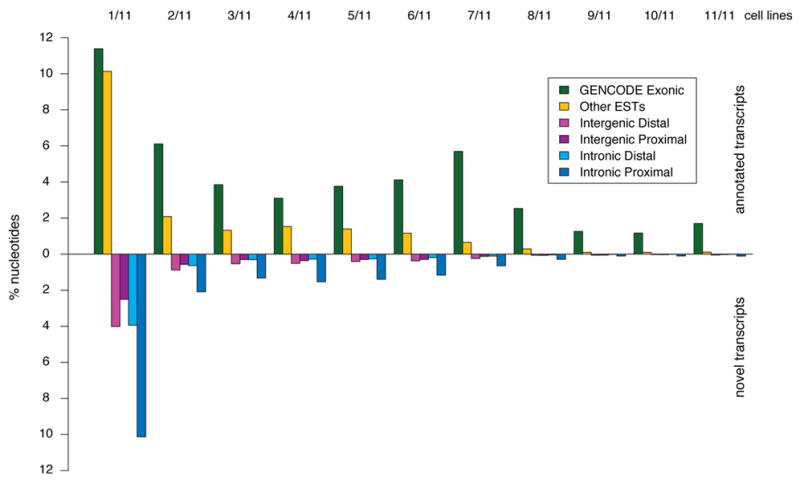
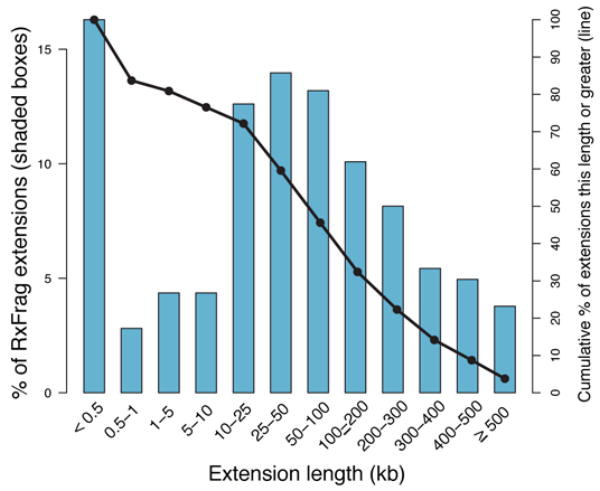
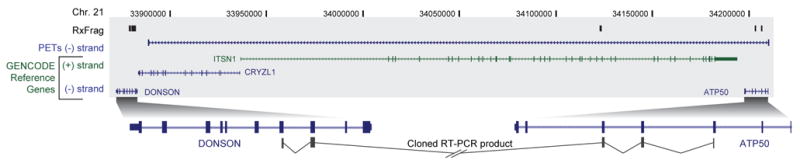
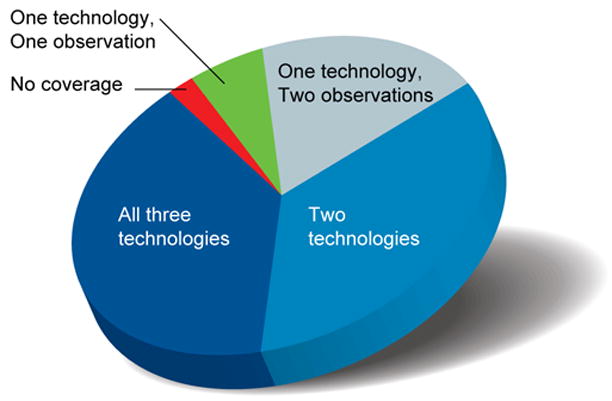
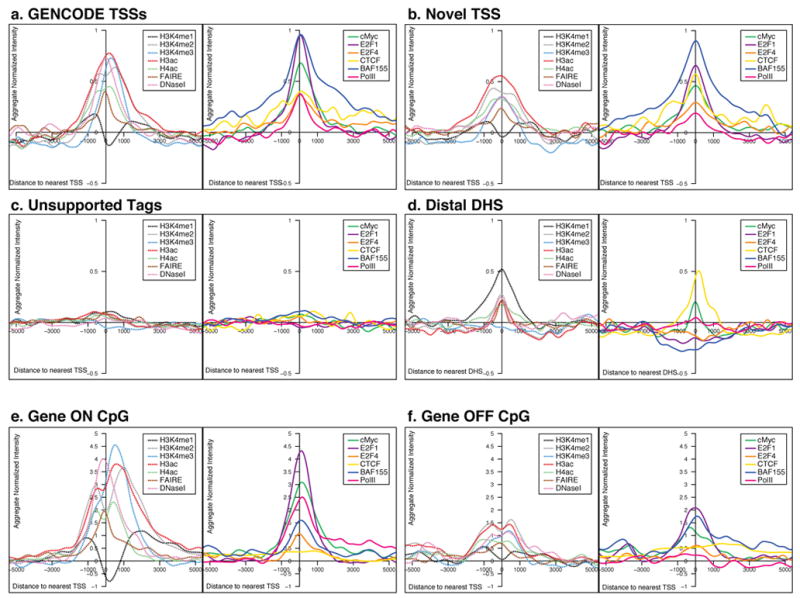
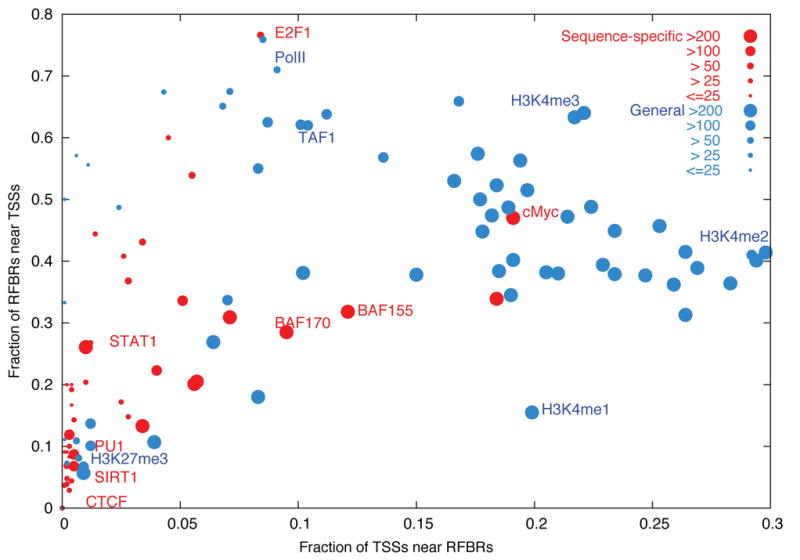
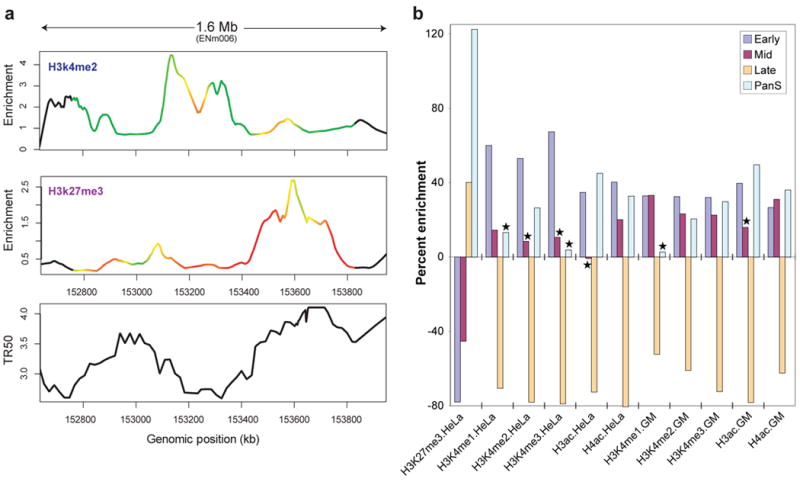
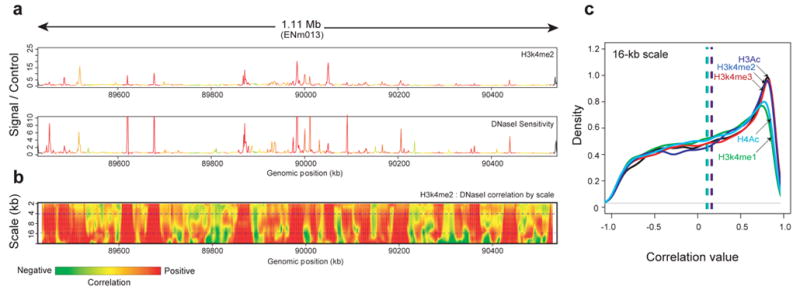
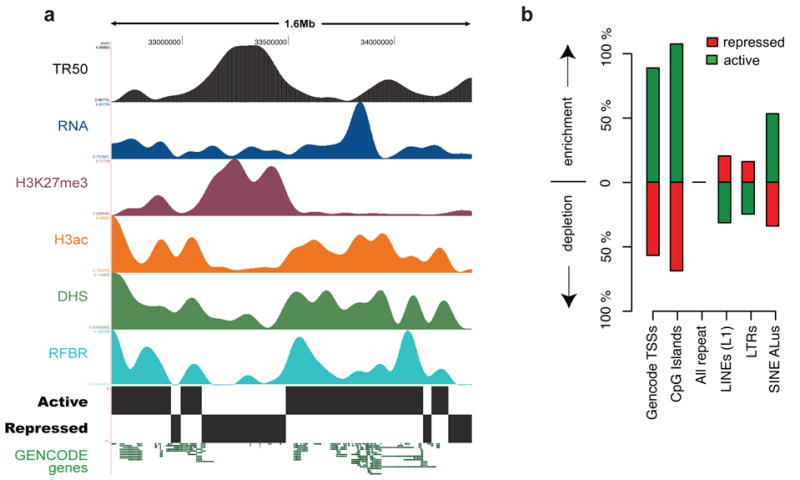
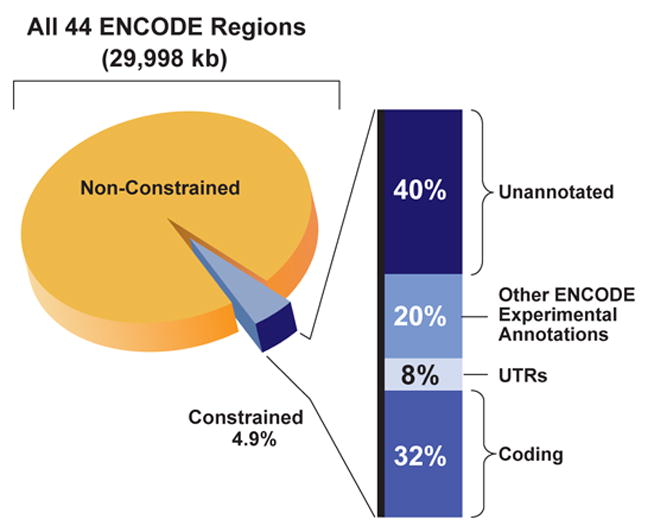
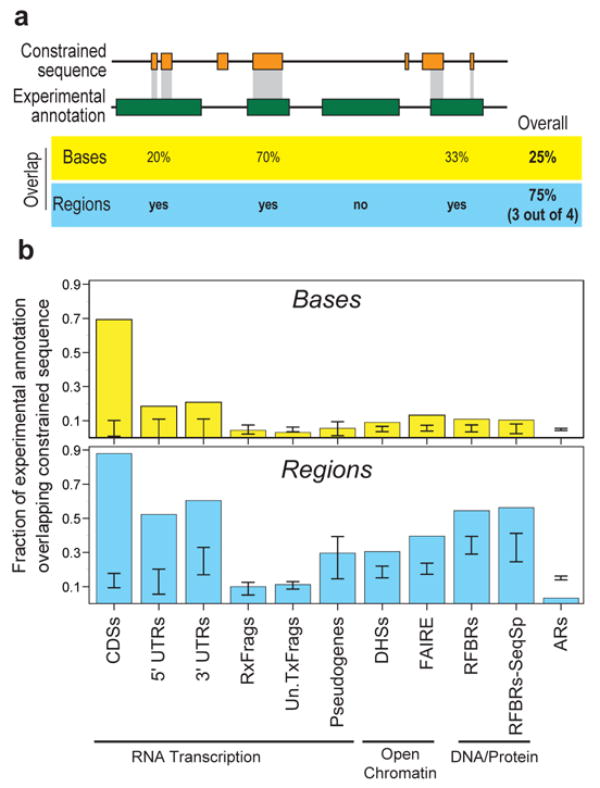
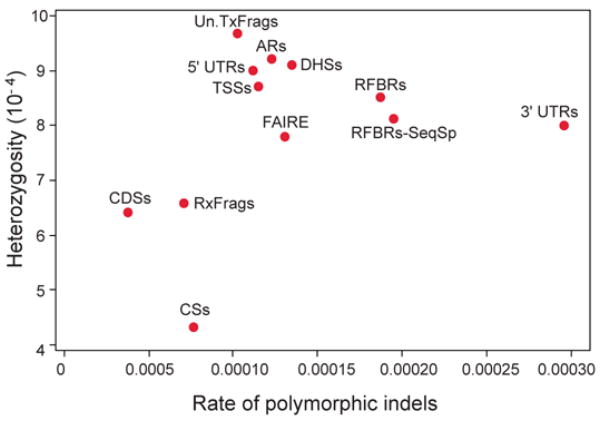
We report the generation and analysis of functional data from multiple, diverse experiments performed on a targeted 1% of the human genome as part of the pilot phase of the ENCODE Project. These data have been further integrated and augmented by a number of evolutionary and computational analyses. Together, our results advance the collective knowledge about human genome function in several major areas. First, our studies provide convincing evidence that the genome is pervasively transcribed, such that the majority of its bases can be found in primary transcripts, including non-protein-coding transcripts, and those that extensively overlap one another. Second, systematic examination of transcriptional regulation has yielded new understanding about transcription start sites, including their relationship to specific regulatory sequences and features of chromatin accessibility and histone modification. Third, a more sophisticated view of chromatin structure has emerged, including its inter-relationship with DNA replication and transcriptional regulation. Finally, integration of these new sources of information, in particular with respect to mammalian evolution based on inter- and intra-species sequence comparisons, has yielded new mechanistic and evolutionary insights concerning the functional landscape of the human genome. Together, these studies are defining a path for pursuit of a more comprehensive characterization of human genome function.
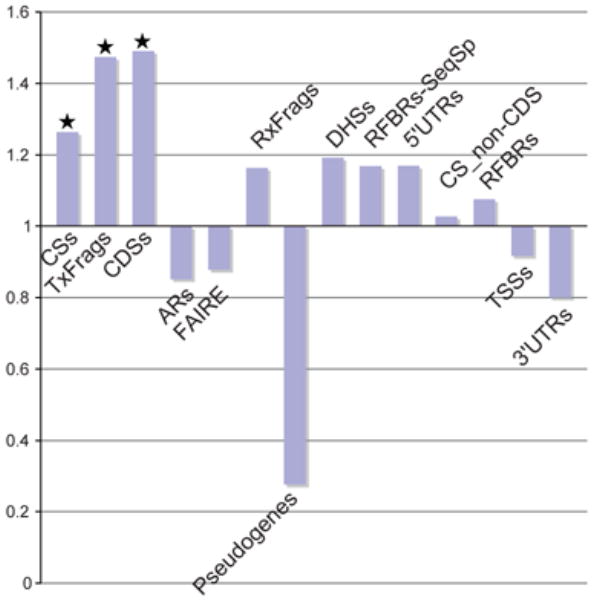
Figures
Comment in
-
Genome project turns up evolutionary surprises.Nature. 2007 Jun 14;447(7146):760-1. doi: 10.1038/447760a. Nature. 2007. PMID: 17568710 No abstract available.
-
Genomics: Encyclopaedia of humble DNA.Nature. 2007 Jun 14;447(7146):782-3. doi: 10.1038/447782a. Nature. 2007. PMID: 17568731 No abstract available.
References
-
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–51. - PubMed
-
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. - PubMed
-
- International Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. - PubMed
-
- Rat Genome Sequencing Project Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HG003541/HG/NHGRI NIH HHS/United States
- U01 HG003147/HG/NHGRI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- R01 HG003143/HG/NHGRI NIH HHS/United States
- P41 HG002371/HG/NHGRI NIH HHS/United States
- U01 HG003156/HG/NHGRI NIH HHS/United States
- R01 HG003532/HG/NHGRI NIH HHS/United States
- F32 CA108313/CA/NCI NIH HHS/United States
- 077198/WT_/Wellcome Trust/United Kingdom
- 062023/WT_/Wellcome Trust/United Kingdom
- R01 HG003110/HG/NHGRI NIH HHS/United States
- U01 HG002523/HG/NHGRI NIH HHS/United States
- U01 HG003161/HG/NHGRI NIH HHS/United States
- R01 HG003521/HG/NHGRI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U01 HG003162/HG/NHGRI NIH HHS/United States
- K22 HG003169/HG/NHGRI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U01 HG003151/HG/NHGRI NIH HHS/United States
- U01 HG003168/HG/NHGRI NIH HHS/United States
- R01 HG003129/HG/NHGRI NIH HHS/United States
- U01 HG003157/HG/NHGRI NIH HHS/United States
- U01 HG003150/HG/NHGRI NIH HHS/United States
- R01 HG002238/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
