

Rho activation regulates CXCL12 chemokine stimulated actin rearrangement and restitution in model intestinal epithelia
- PMID: 17572689
- PMCID: PMC2693067
- DOI: 10.1038/labinvest.3700595
Rho activation regulates CXCL12 chemokine stimulated actin rearrangement and restitution in model intestinal epithelia
Abstract
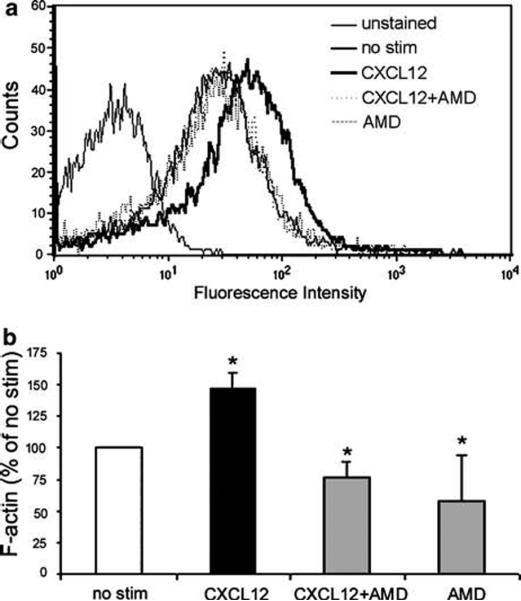
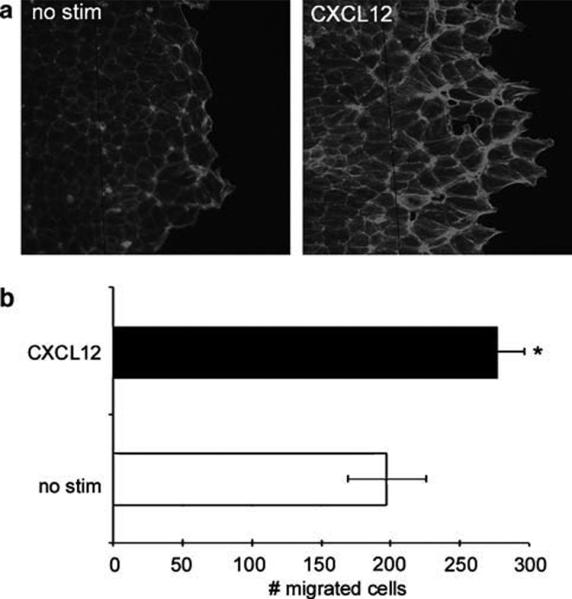
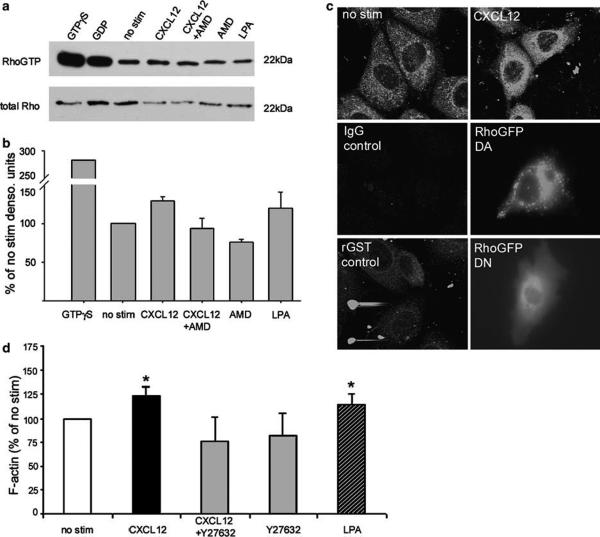
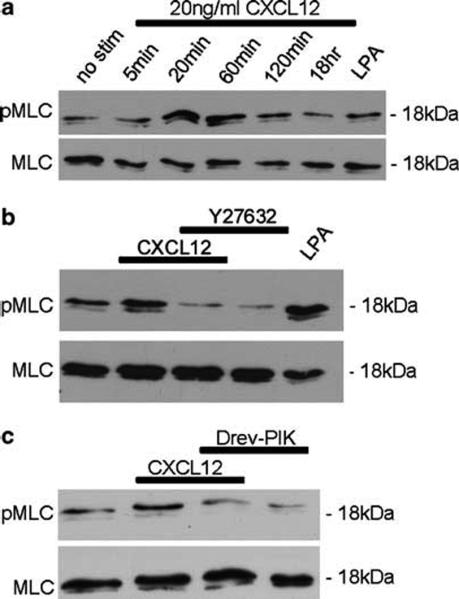
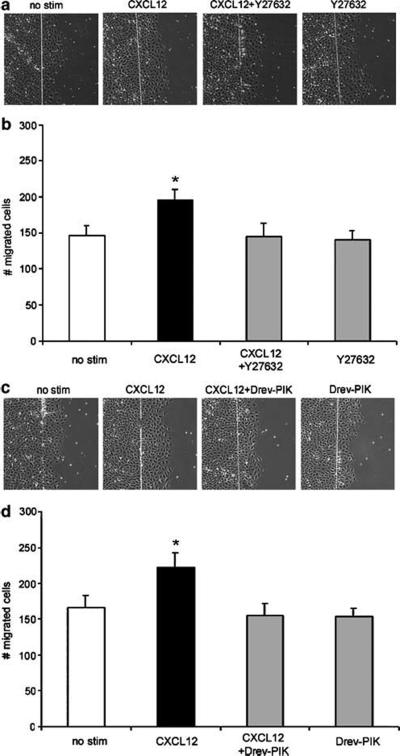
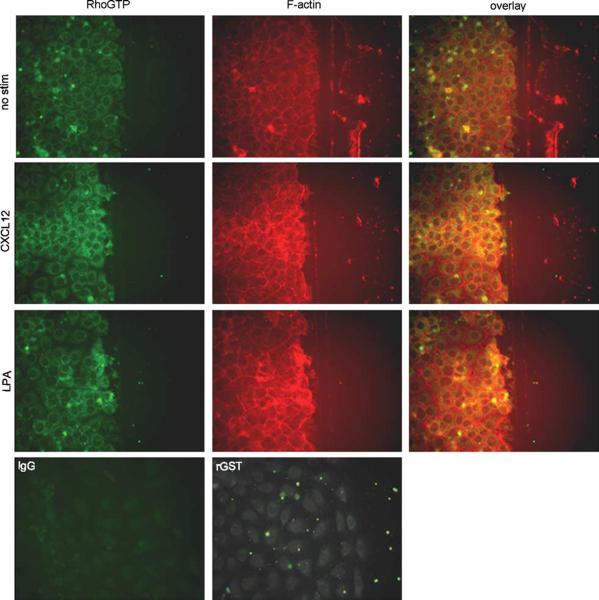
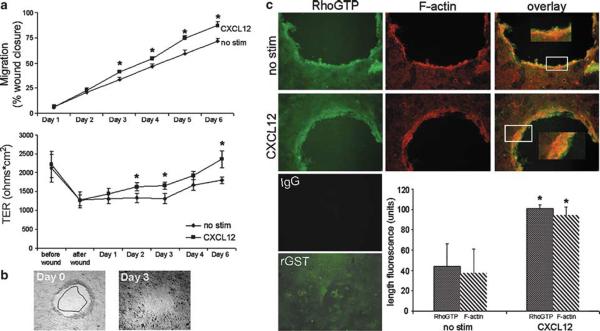
Chemokines are critical regulatory factors that direct migration, proliferation and maturation of receptor expressing target cells within gut mucosa. The aim of the present study was to define the cellular mechanisms whereby engagement of the essential chemokine CXCL12 to CXCR4 regulates restitutive epithelial cell migration. Non-transformed IEC-6 cells or polarized T84 epithelial monolayers were wounded and F-actin accumulation assessed using fluorescence microscopy and flow cytometry. Immunoblot analysis, pull-down assays, fluorescence microscopy and wound healing assays defined activation of Rho, Rho-kinase (ROCK), and myosin light chain (MLC) and the role for those Rho effectors in CXCL12-regulated epithelial restitution. CXCL12 increased RhoGTP and F-actin localization to the leading edge of wounded IEC-6 and T84 monolayers. CXCL12 congruently stimulated an increase in active MLC that was inhibited by blockade of ROCK and myosin light chain kinase and regulated epithelial migration. Our data in model intestinal epithelia suggest CXCR4 and CXCL12 may function as an autocrine and paracrine mucosal signaling network regulating the competency of the epithelial barrier to withstand injury and mediate repair following damage.
Figures
References
-
- Booth C, Brady G, Potten CS. Crowd control in the crypt. Nat Med. 2002;8:1360–1361. - PubMed
-
- Mammen JM, Matthews JB. Mucosal repair in the gastrointestinal tract. Crit Care Med. 2003;31:S532–S537. - PubMed
-
- Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7:68–77. - PubMed
-
- Jacinto A, Martinez-Arias A, Martin P. Mechanisms of epithelial fusion and repair. Nat Cell Biol. 2001;3:E117–E123. - PubMed
-
- Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
