

Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway
- PMID: 17575307
- PMCID: PMC2085232
- DOI: 10.1093/hmg/ddm141
Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway
Abstract
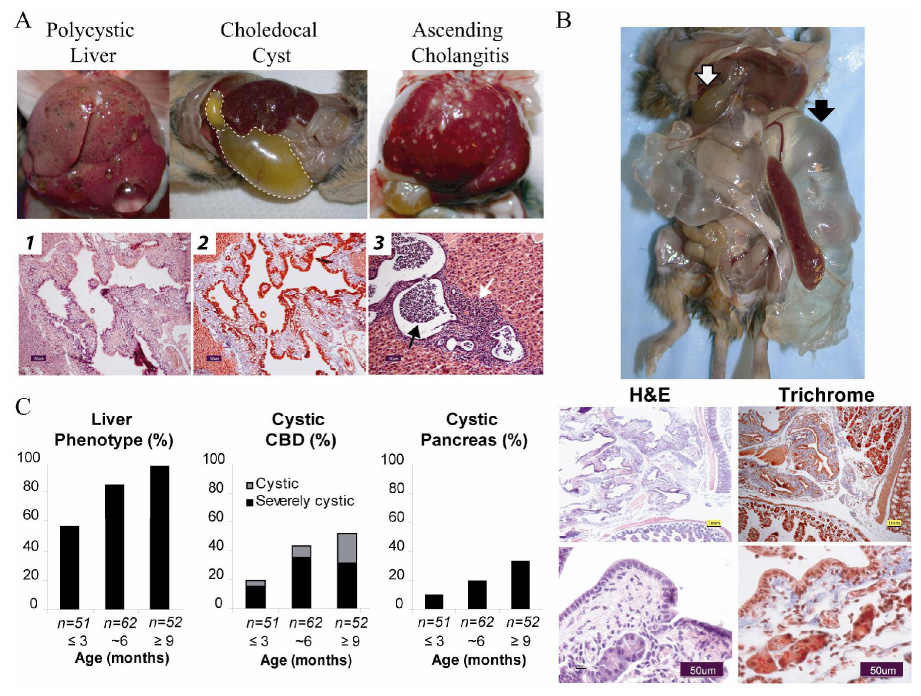
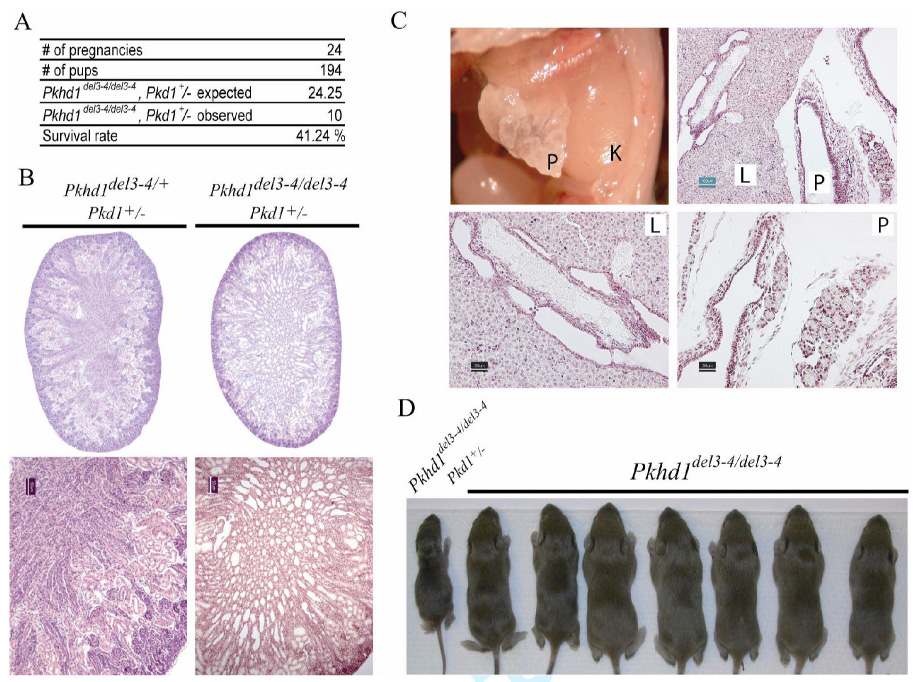
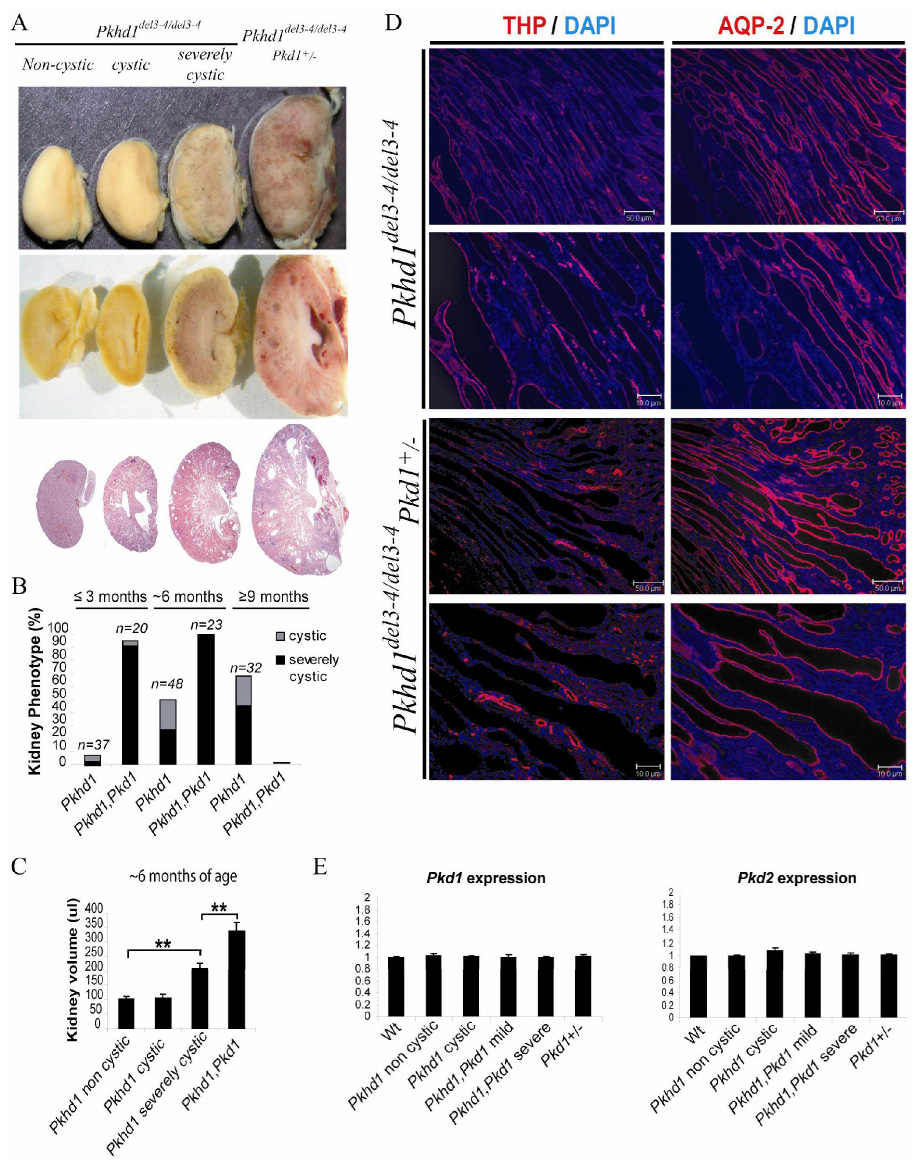
Polycystic kidney disease (PKD) describes a heterogeneous collection of disorders that differ significantly with respect to their etiology and clinical presentation. They share, however, abnormal tubular morphology as a common feature, leading to the hypothesis that their respective gene products may function cooperatively in a common pathway to maintain tubular integrity. To study the pathobiology of one major form of human PKD, we generated a mouse line with a floxed allele of Pkhd1, the orthologue of the gene mutated in human autosomal recessive PKD. Cre-mediated excision of exons 3-4 results in a probable hypomorphic allele. Pkhd1(del3-4/del3-4) developed a range of phenotypes that recapitulate key features of the human disease. Like in humans, abnormalities of the biliary tract were an invariant finding. Most mice 6 months or older also developed renal cysts. Subsets of animals presented with either perinatal respiratory failure or exhibited growth retardation that was not due to the renal disease. We then tested for genetic interaction between Pkhd1 and Pkd1, the mouse orthologue of the gene most commonly linked to human autosomal dominant PKD. Pkd1(+/-); Pkhd1(del3-4/del3-4) mice had markedly more severe disease than Pkd1(+/+); Pkhd1(del3-4/del3-4) littermates. These studies are the first to show genetic interaction between the major loci responsible for human renal cystic disease in a common PKD pathway.
Figures



References
-
- Gabow PA. Autosomal Dominant Polycystic Kidney Disease. 1993;329:332–342. - PubMed
-
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1995;5:2048–2056. - PubMed
-
- The European Polycystic Kidney Disease Consortium. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77:881–894. - PubMed
-
- Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
