

Epigenetic inheritance in rice plants
- PMID: 17576658
- PMCID: PMC2735323
- DOI: 10.1093/aob/mcm110
Epigenetic inheritance in rice plants
Abstract
Background and aims: Epigenetics is defined as mechanisms that regulate gene expression without base sequence alteration. One molecular basis is considered to be DNA cytosine methylation, which reversibly modifies DNA or chromatin structures. Although its correlation with epigenetic inheritance over generations has been circumstantially shown, evidence at the gene level has been limited. The present study aims to find genes whose methylation status directly correlates with inheritance of phenotypic changes.
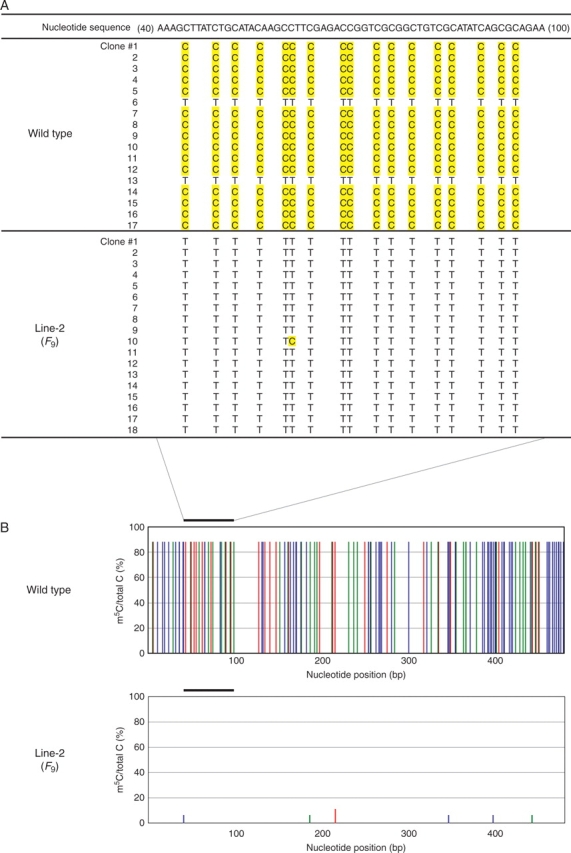
Methods: DNA methylation in vivo was artificially reduced by treating rice (Oryza sativa ssp. japonica) seeds with 5-azadeoxycytidine, and the progeny were cultivated in the field for > 10 years. Genomic regions with changed methylation status were screened by the methylation-sensitive amplified polymorphysm (MSAP) method, and cytosine methylation was directly scanned by the bisulfite mapping method. Pathogen infection with Xanthomonas oryzae pv. oryzae, race PR2 was performed by the scissors-dip method on mature leaf blades.
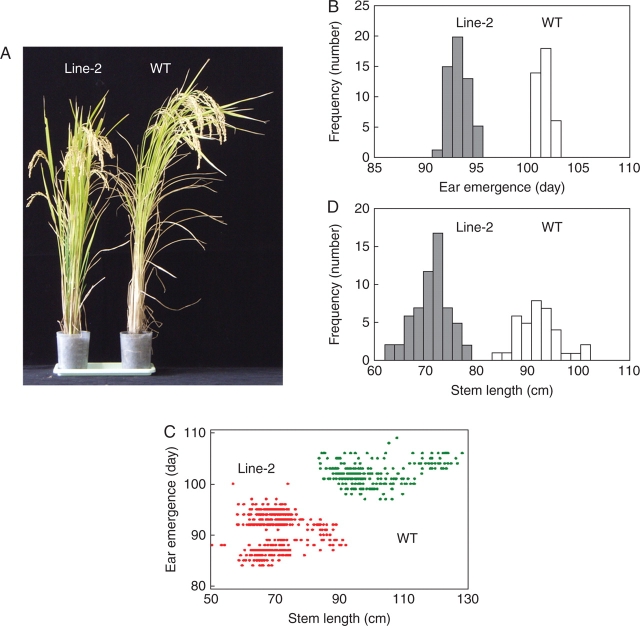
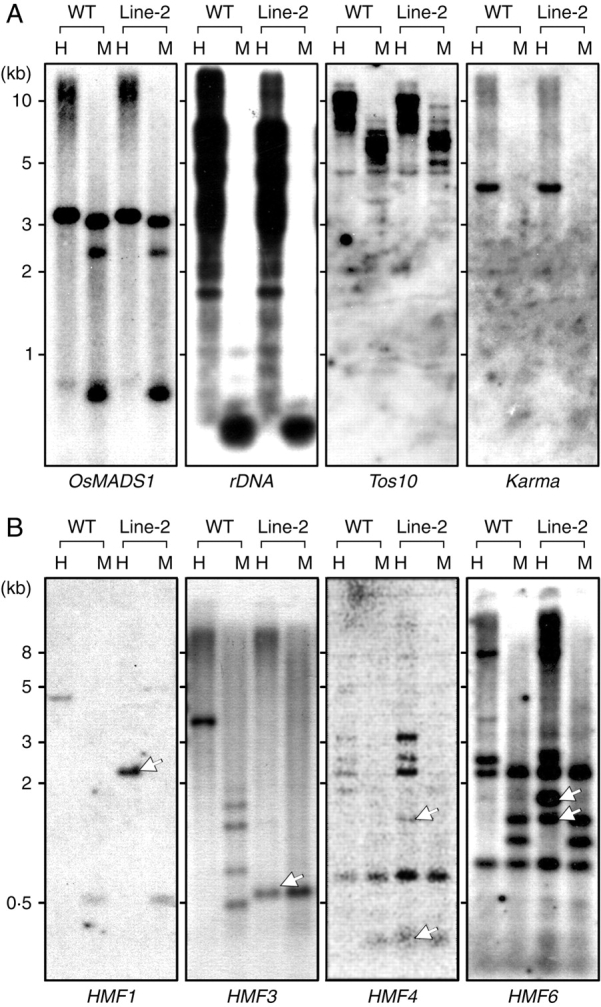
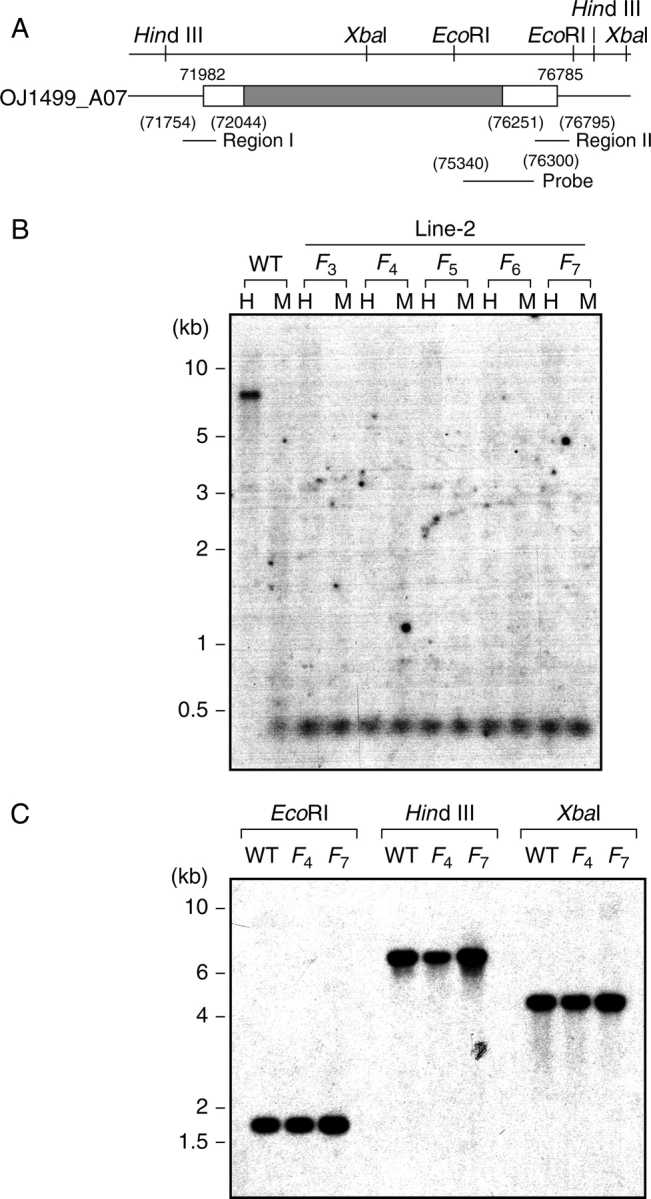
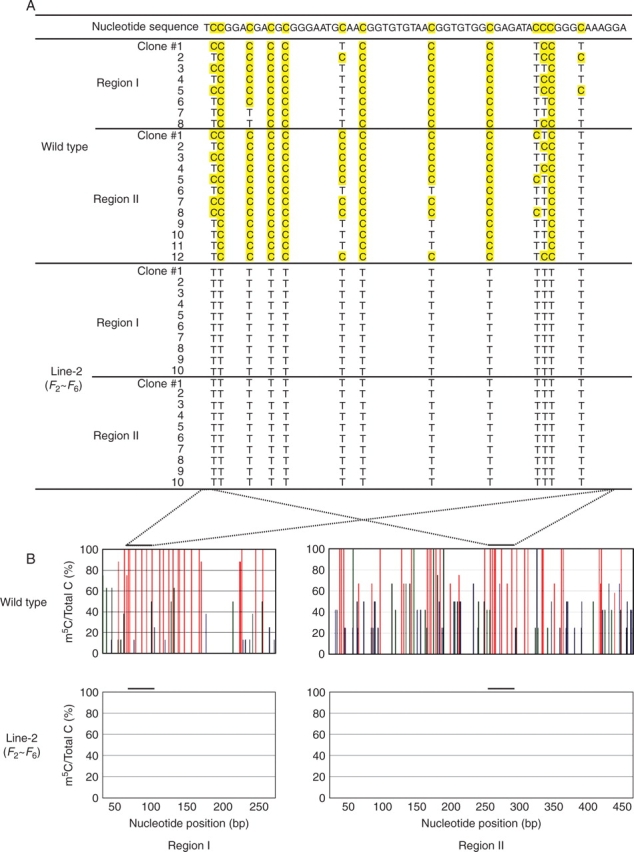
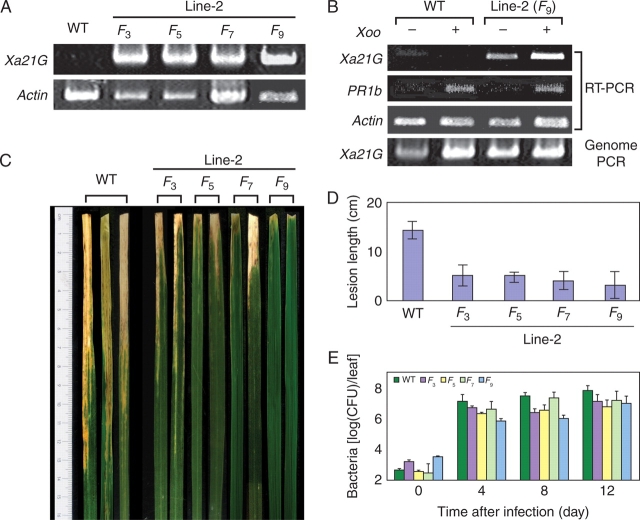
Key results: The majority of seedlings were lethal, but some survived to maturity. One line designated as Line-2 showed a clear marker phenotype of dwarfism, which was stably inherited by the progeny over nine generations. MSAP screening identified six fragments, among which two were further characterized by DNA blot hybridization and direct methylation mapping. One clone encoding a retrotransposon gag-pol polyprotein showed a complete erasure of 5-methylcytosines in Line-2, but neither translocation nor expression of this region was detectable. The other clone encoded an Xa21-like protein, Xa21G. In wild-type plants, all cytosines were methylated within the promoter region, whereas in Line-2, corresponding methylation was completely erased throughout generations. Expression of Xa21G was not detectable in wild type but was constitutive in Line-2. When infected with X. oryzae pv. oryzae, against which Xa21 confers resistance in a gene-for-gene manner, the progeny of Line-2 were apparently resistant while the wild type was highly susceptible without Xa21G expression.
Conclusions: These results indicated that demethylation was selective in Line-2, and that promoter demethylation abolished the constitutive silencing of Xa21G due to hypermethylation, resulting in acquisition of disease resistance. Both hypomethylation and resistant trait were stably inherited. This is a clear example of epigenetic inheritance, and supports the idea of Lamarckian inheritance which suggested acquired traits to be heritable.
Figures

References
-
- Aina R, Sgorbati S, Santagostino A, Labra M, Ghiani A, Citterio S. Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp. Physiologia Plantarum. 2004;121:472–480.
-
- Amado L, Abranches R, Neves N, Viegas W. Development-dependent inheritance of 5-azacytidine-induced epimutations in triticale: analysis of rDNA expression patterns. Chromosome Research. 1997;5:445–450. - PubMed
-
- Bender J. DNA methylation and epigenetics. Annual Review of Plant Biology. 2004;55:41–68. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes and Development. 2002;16:6–21. - PubMed
-
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nature Genetics. 2006;38:626–635. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
