

A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets
- PMID: 17576779
- PMCID: PMC2118641
- DOI: 10.1084/jem.20070058
A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets
Abstract
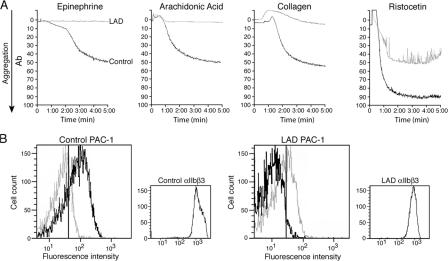
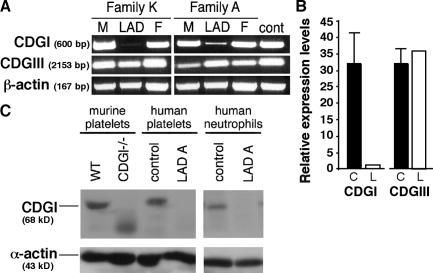
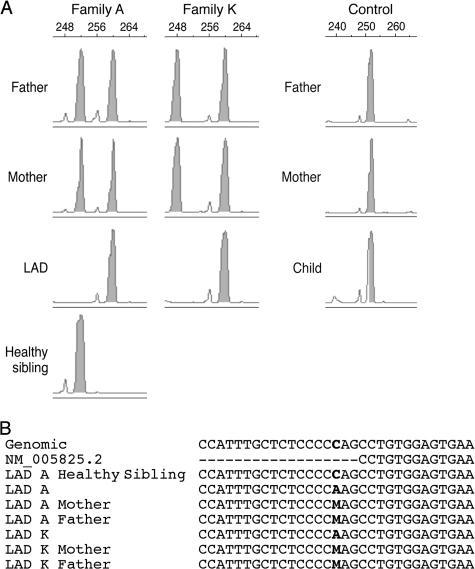
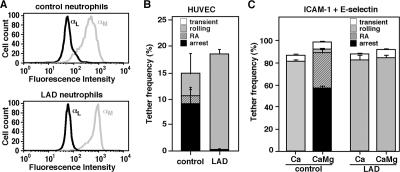
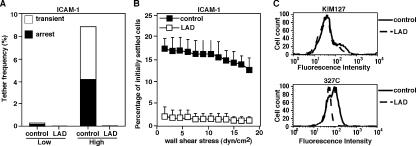
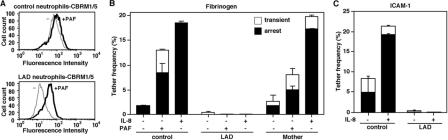
Leukocyte and platelet integrins rapidly alter their affinity and adhesiveness in response to various activation (inside-out) signals. A rare leukocyte adhesion deficiency (LAD), LAD-III, is associated with severe defects in leukocyte and platelet integrin activation. We report two new LAD cases in which lymphocytes, neutrophils, and platelets share severe defects in beta(1), beta(2), and beta(3) integrin activation. Patients were both homozygous for a splice junction mutation in their CalDAG-GEFI gene, which is a key Rap-1/2 guanine exchange factor (GEF). Both mRNA and protein levels of the GEF were diminished in LAD lymphocytes, neutrophils, and platelets. Consequently, LAD-III platelets failed to aggregate because of an impaired alpha(IIb)beta(3) activation by key agonists. beta(2) integrins on LAD-III neutrophils were unable to mediate leukocyte arrest on TNFalpha-stimulated endothelium, despite normal selectin-mediated rolling. In situ subsecond activation of neutrophil beta(2) integrin adhesiveness by surface-bound chemoattractants and of primary T lymphocyte LFA-1 by the CXCL12 chemokine was abolished. Chemokine inside-out signals also failed to stimulate lymphocyte LFA-1 extension and high affinity epitopes. Chemokine-triggered VLA-4 adhesiveness in T lymphocytes was partially defective as well. These studies identify CalDAG-GEFI as a critical regulator of inside-out integrin activation in human T lymphocytes, neutrophils, and platelets.
Figures

References
-
- Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 76:301–314. - PubMed
-
- Carman, C.V., and T.A. Springer. 2003. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin. Cell Biol. 15:547–556. - PubMed
-
- Campbell, J.J., and E.C. Butcher. 2000. Chemokines in tissue-specific and microenvironment-specific lymphocyte homing. Curr. Opin. Immunol. 12:336–341. - PubMed
-
- Ley, K. 2003. Arrest chemokines. Microcirculation. 10:289–295. - PubMed
-
- Ginsberg, M.H., A. Partridge, and S.J. Shattil. 2005. Integrin regulation. Curr. Opin. Cell Biol. 17:509–516. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
