

Epigenetic natural variation in Arabidopsis thaliana
- PMID: 17579518
- PMCID: PMC1892575
- DOI: 10.1371/journal.pbio.0050174
Epigenetic natural variation in Arabidopsis thaliana
Abstract
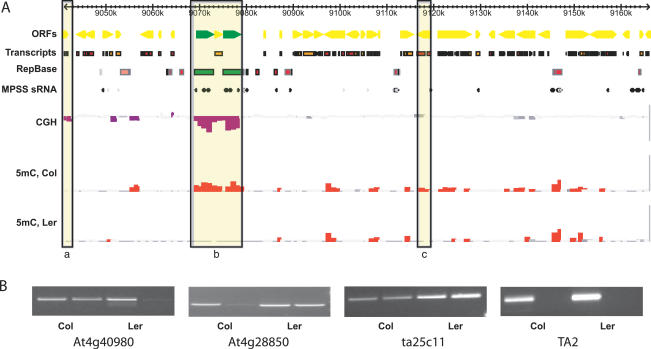
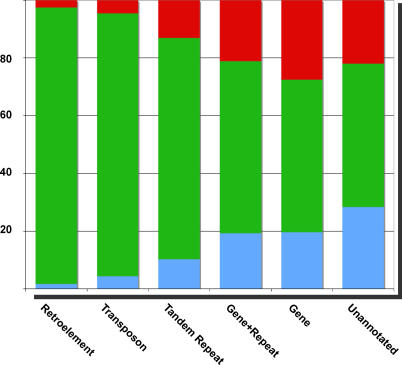
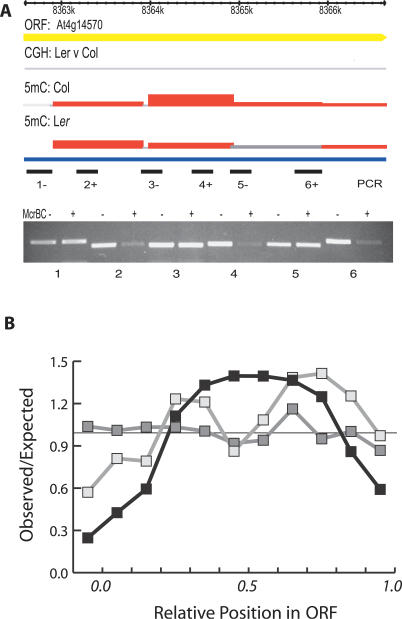
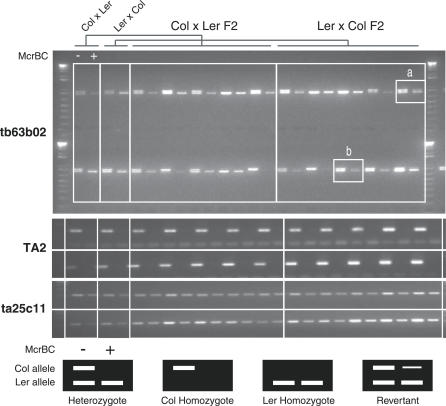
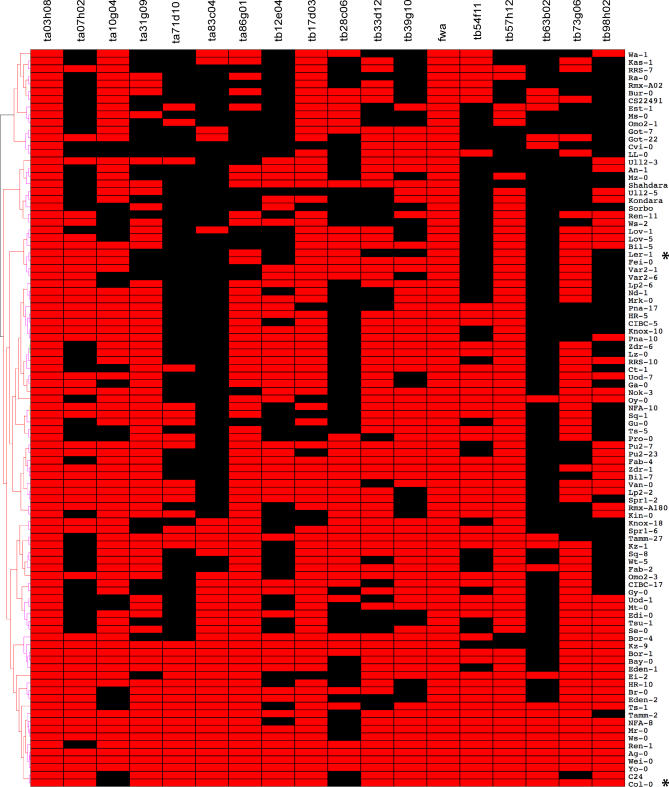
Cytosine methylation of repetitive sequences is widespread in plant genomes, occurring in both symmetric (CpG and CpNpG) as well as asymmetric sequence contexts. We used the methylation-dependent restriction enzyme McrBC to profile methylated DNA using tiling microarrays of Arabidopsis Chromosome 4 in two distinct ecotypes, Columbia and Landsberg erecta. We also used comparative genome hybridization to profile copy number polymorphisms. Repeated sequences and transposable elements (TEs), especially long terminal repeat retrotransposons, are densely methylated, but one third of genes also have low but detectable methylation in their transcribed regions. While TEs are almost always methylated, genic methylation is highly polymorphic, with half of all methylated genes being methylated in only one of the two ecotypes. A survey of loci in 96 Arabidopsis accessions revealed a similar degree of methylation polymorphism. Within-gene methylation is heritable, but is lost at a high frequency in segregating F(2) families. Promoter methylation is rare, and gene expression is not generally affected by differences in DNA methylation. Small interfering RNA are preferentially associated with methylated TEs, but not with methylated genes, indicating that most genic methylation is not guided by small interfering RNA. This may account for the instability of gene methylation, if occasional failure of maintenance methylation cannot be restored by other means.
Conflict of interest statement
Figures
References
-
- Bender J. DNA methylation and epigenetics. Annu Rev Plant Biol. 2004;55:41–68. - PubMed
-
- Chan SW, Henderson IR, Jacobsen SE. Gardening the genome: DNA methylation in Arabidopsis thaliana . Nat Rev Genet. 2005;6:351–360. - PubMed
-
- Klose RJ, Bird AP. Genomic DNA methylation: The mark and its mediators. Trends Biochem Sci. 2006;31:89–97. - PubMed
-
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
