

Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration
- PMID: 17579875
- PMCID: PMC2827877
- DOI: 10.1007/s00401-007-0237-2
Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration
Abstract
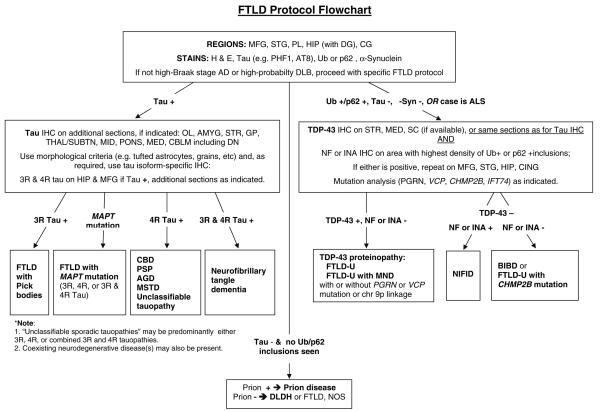
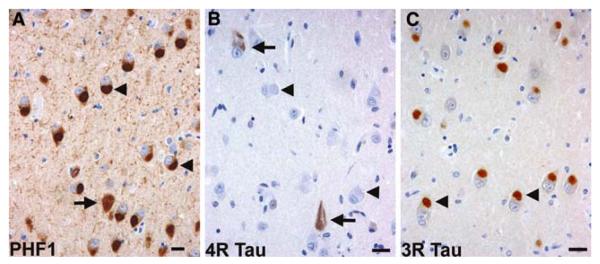
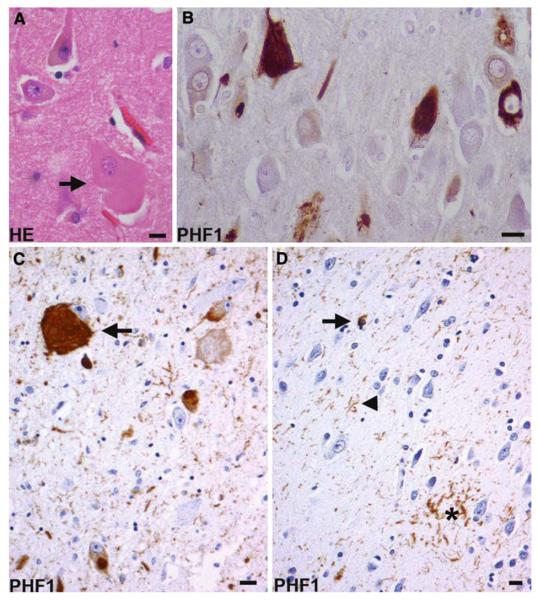
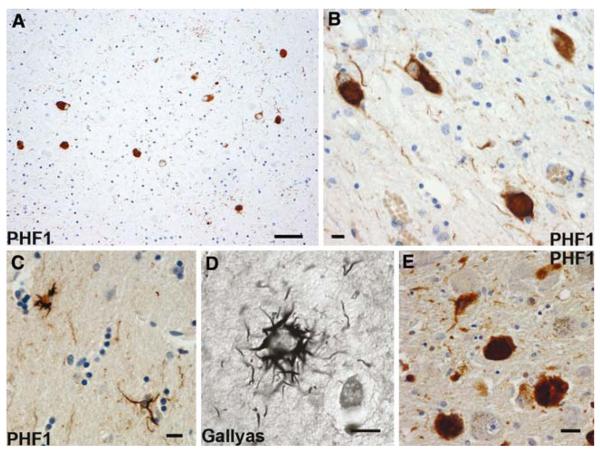
The aim of this study was to improve the neuropathologic recognition and provide criteria for the pathological diagnosis in the neurodegenerative diseases grouped as frontotemporal lobar degeneration (FTLD); revised criteria are proposed. Recent advances in molecular genetics, biochemistry, and neuropathology of FTLD prompted the Midwest Consortium for Frontotemporal Lobar Degeneration and experts at other centers to review and revise the existing neuropathologic diagnostic criteria for FTLD. The proposed criteria for FTLD are based on existing criteria, which include the tauopathies [FTLD with Pick bodies, corticobasal degeneration, progressive supranuclear palsy, sporadic multiple system tauopathy with dementia, argyrophilic grain disease, neurofibrillary tangle dementia, and FTD with microtubule-associated tau (MAPT) gene mutation, also called FTD with parkinsonism linked to chromosome 17 (FTDP-17)]. The proposed criteria take into account new disease entities and include the novel molecular pathology, TDP-43 proteinopathy, now recognized to be the most frequent histological finding in FTLD. TDP-43 is a major component of the pathologic inclusions of most sporadic and familial cases of FTLD with ubiquitin-positive, tau-negative inclusions (FTLD-U) with or without motor neuron disease (MND). Molecular genetic studies of familial cases of FTLD-U have shown that mutations in the progranulin (PGRN) gene are a major genetic cause of FTLD-U. Mutations in valosin-containing protein (VCP) gene are present in rare familial forms of FTD, and some families with FTD and/or MND have been linked to chromosome 9p, and both are types of FTLD-U. Thus, familial TDP-43 proteinopathy is associated with defects in multiple genes, and molecular genetics is required in these cases to correctly identify the causative gene defect. In addition to genetic heterogeneity amongst the TDP-43 proteinopathies, there is also neuropathologic heterogeneity and there is a close relationship between genotype and FTLD-U subtype. In addition to these recent significant advances in the neuropathology of FTLD-U, novel FTLD entities have been further characterized, including neuronal intermediate filament inclusion disease. The proposed criteria incorporate up-to-date neuropathology of FTLD in the light of recent immunohistochemical, biochemical, and genetic advances. These criteria will be of value to the practicing neuropathologist and provide a foundation for clinical, clinico-pathologic, mechanistic studies and in vivo models of pathogenesis of FTLD.
Figures
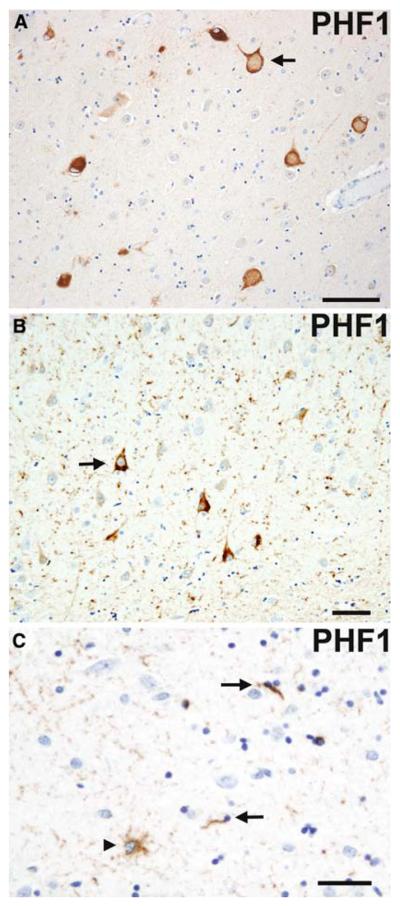
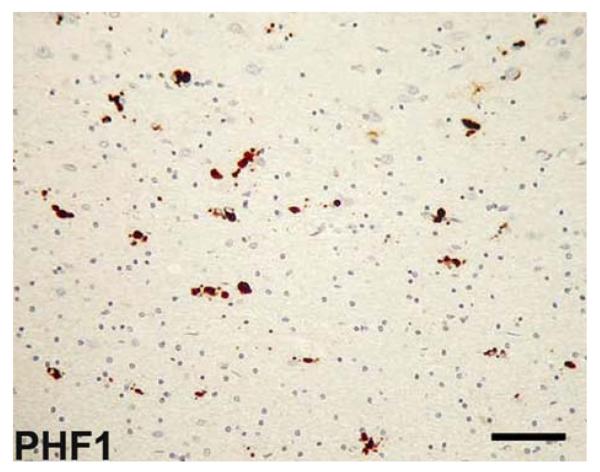

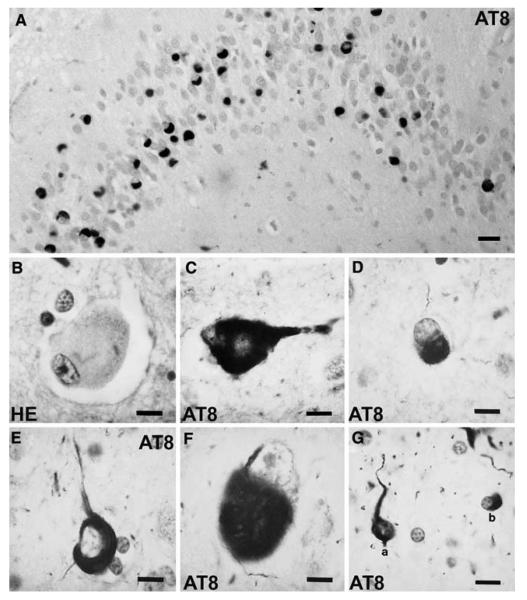
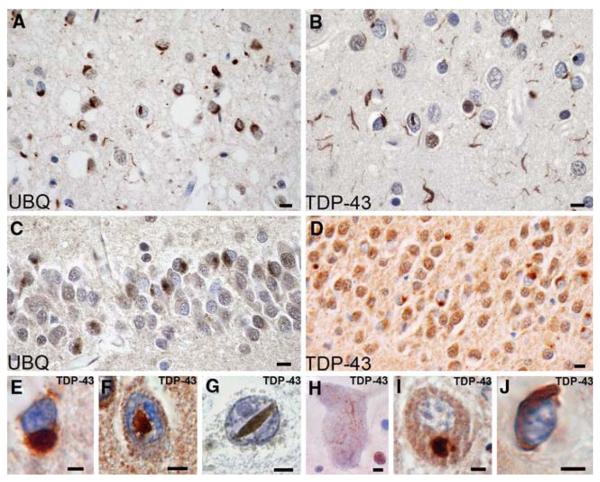
References
-
- Alafuzoff I, Pikkarainen M, Al Sarraj S, Arzberger T, Bell J, Bodi I, Bogdanovic N, Budka H, Bugiani O, Ferrer I, Gelpi E, Giaccone G, Graeber MB, Hauw JJ, Kamphorst W, King A, Kopp N, Korkolopoulou P, Kovacs GG, Meyronet D, Parchi P, Patsouris E, Preusser M, Ravid R, Roggendorf W, Seilhean D, Streichenberger N, Thal DR, Kretzschmar H. Interlaboratory comparison of assessments of Alzheimer disease-related lesions: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol. 2006;65:740–757. - PubMed
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. - PubMed
-
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. - PubMed
-
- Behrens MI, Mukherjee O, Tu PH, Liscic RM, Grinberg LT, Carter D, Paulsmeyer K, Taylor-Reinwald L, Gitcho M, Norton JB, Chakraverty S, Goate AM, Morris JC, Cairns NJ. Neuropathologic heterogeneity in HDDD1: a familial frontotemporal lobar degeneration with ubiquitin-positive inclusions and progranulin mutation. Alzheimer Dis Assoc Disord. 2007;21:1–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 AG013854/AG/NIA NIH HHS/United States
- AG10124/AG/NIA NIH HHS/United States
- P01 AG017586/AG/NIA NIH HHS/United States
- U01-AG16976/AG/NIA NIH HHS/United States
- AG17586/AG/NIA NIH HHS/United States
- P30-AG13854/AG/NIA NIH HHS/United States
- P30-NS057105/NS/NINDS NIH HHS/United States
- P50-AG05681/AG/NIA NIH HHS/United States
- P01-AG03991/AG/NIA NIH HHS/United States
- P30 NS057105/NS/NINDS NIH HHS/United States
- P30 AG010124/AG/NIA NIH HHS/United States
- U01 AG016976/AG/NIA NIH HHS/United States
- P01 AG003991/AG/NIA NIH HHS/United States
- P50 AG005681/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
