

Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes
- PMID: 17586620
- PMCID: PMC2085107
- DOI: 10.1152/ajpheart.00355.2007
Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes
Abstract
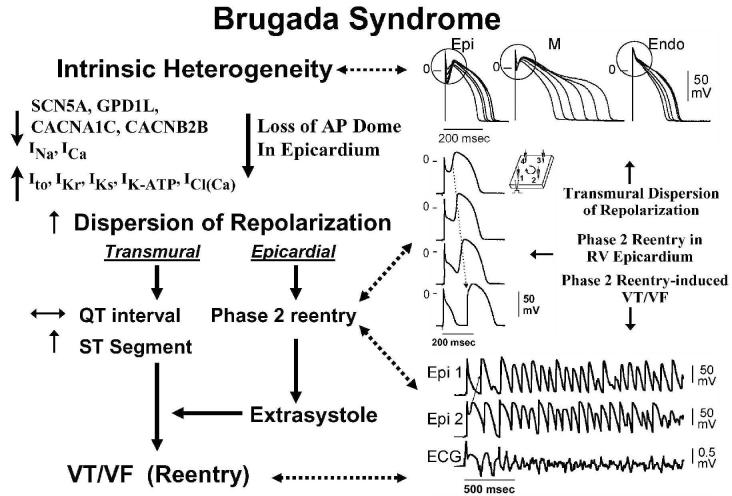
This review examines the role of spatial electrical heterogeneity within the ventricular myocardium on the function of the heart in health and disease. The cellular basis for transmural dispersion of repolarization (TDR) is reviewed, and the hypothesis that amplification of spatial dispersion of repolarization underlies the development of life-threatening ventricular arrhythmias associated with inherited ion channelopathies is evaluated. The role of TDR in long QT, short QT, and Brugada syndromes, as well as catecholaminergic polymorphic ventricular tachycardia (VT), is critically examined. In long QT syndrome, amplification of TDR is often secondary to preferential prolongation of the action potential duration (APD) of M cells; in Brugada syndrome, however, it is thought to be due to selective abbreviation of the APD of the right ventricular epicardium. Preferential abbreviation of APD of the endocardium or epicardium appears to be responsible for the amplification of TDR in short QT syndrome. In catecholaminergic polymorphic VT, reversal of the direction of activation of the ventricular wall is responsible for the increase in TDR. In conclusion, long QT, short QT, Brugada, and catecholaminergic polymorphic VT syndromes are pathologies with very different phenotypes and etiologies, but they share a common final pathway in causing sudden cardiac death.
Figures








References
-
- Aiba T, Shimizu W, Hidaka I, Uemura K, Noda T, Zheng C, Kamiya A, Inagaki M, Sugimachi M, Sunagawa K. Cellular basis for trigger and maintenance of ventricular fibrillation in the Brugada syndrome model: high-resolution optical mapping study. J Am Coll Cardiol. 2006;47:2074–2085. - PubMed
-
- Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–645. - PubMed
-
- Akar FG, Tomaselli GF. Conduction abnormalities in nonischemic dilated cardiomyopathy: basic mechanisms and arrhythmic consequences. Trends Cardiovasc Med. 2005;15:259–264. - PubMed
-
- Antzelevitch C. The Brugada Syndrome: Diagnostic Criteria and Cellular Mechanisms. Eur Heart J. 2001;22:356–363. - PubMed
-
- Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268–272. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources