

Development of a gene knockout system in Candida parapsilosis reveals a conserved role for BCR1 in biofilm formation
- PMID: 17586721
- PMCID: PMC1951126
- DOI: 10.1128/EC.00136-07
Development of a gene knockout system in Candida parapsilosis reveals a conserved role for BCR1 in biofilm formation
Abstract
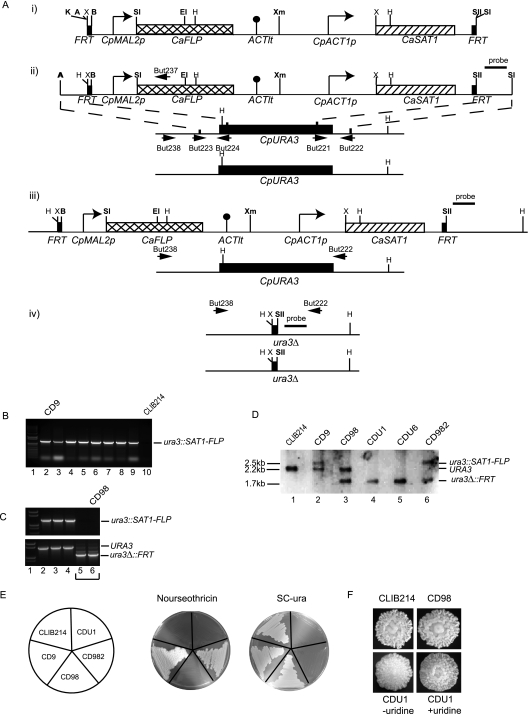
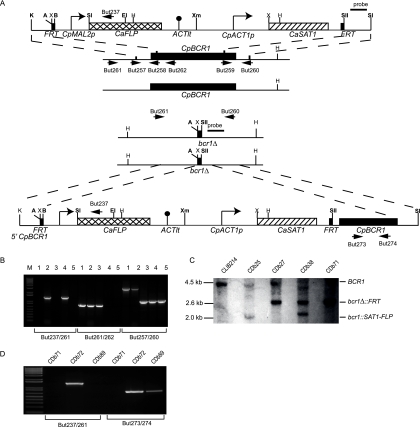
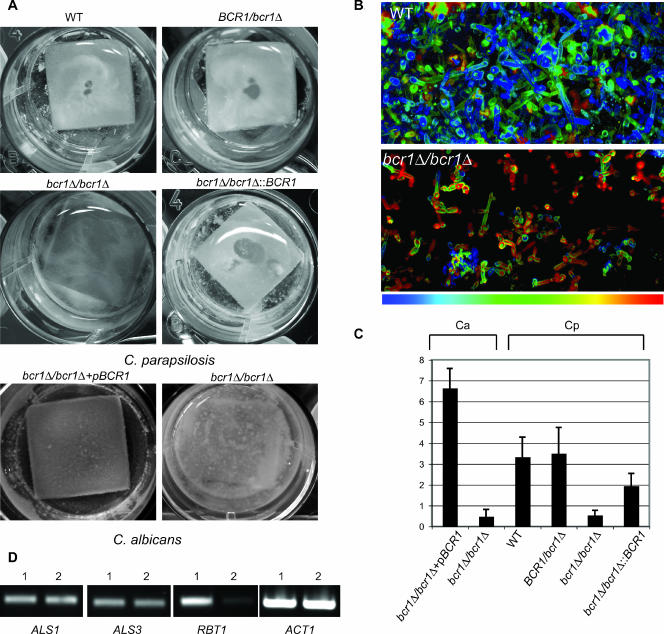
Candida parapsilosis is an important cause of candidiasis, yet few molecular tools are available. We adapted a recyclable nourseothricin resistance marker gene (SAT1) originally developed for use with C. albicans in order to generate gene knockouts from C. parapsilosis. We first replaced the promoters driving expression of the FLP recombinase and the SAT1 genes with the equivalent sequences from C. parapsilosis. We then used the cassette to generate a homozygous knockout of C. parapsilosis URA3. The ura3 knockouts have altered colony morphologies. We also knocked out both alleles of an ortholog of BCR1, a gene encoding a transcription factor known to be required for biofilm development in C. albicans. We show that C. parapsilosis BCR1 is necessary for biofilm development in C. parapsilosis and for expression of the cell wall protein encoded by RBT1. Our results suggest that there are significant similarities in the regulation of biofilms between the two species, despite the fact that C. parapsilosis does not generate true hyphae and that BCR1 regulates the expression of many hypha-specific adhesins in C. albicans.
Figures
Similar articles
-
Biofilm formation and genetic variability of BCR1 gene in the Candida parapsilosis complex.Rev Iberoam Micol. 2015 Jul-Sep;32(3):180-4. doi: 10.1016/j.riam.2014.11.001. Epub 2015 Feb 20. Rev Iberoam Micol. 2015. PMID: 25843001
-
The SAT1 flipper, an optimized tool for gene disruption in Candida albicans.Gene. 2004 Oct 27;341:119-27. doi: 10.1016/j.gene.2004.06.021. Gene. 2004. PMID: 15474295
-
Conserved and divergent roles of Bcr1 and CFEM proteins in Candida parapsilosis and Candida albicans.PLoS One. 2011;6(12):e28151. doi: 10.1371/journal.pone.0028151. Epub 2011 Dec 1. PLoS One. 2011. PMID: 22145027 Free PMC article.
-
Adherence and biofilm formation of non-Candida albicans Candida species.Trends Microbiol. 2011 May;19(5):241-7. doi: 10.1016/j.tim.2011.02.003. Epub 2011 Mar 15. Trends Microbiol. 2011. PMID: 21411325 Review.
-
Promoter regulation in Candida albicans and related species.FEMS Yeast Res. 2009 Feb;9(1):2-15. doi: 10.1111/j.1567-1364.2008.00455.x. Epub 2008 Nov 12. FEMS Yeast Res. 2009. PMID: 19054124 Review.
Cited by
-
Comparative evolution of morphological regulatory functions in Candida species.Eukaryot Cell. 2013 Oct;12(10):1356-68. doi: 10.1128/EC.00164-13. Epub 2013 Aug 2. Eukaryot Cell. 2013. PMID: 23913541 Free PMC article.
-
Evolution of mating within the Candida parapsilosis species group.Eukaryot Cell. 2011 Apr;10(4):578-87. doi: 10.1128/EC.00276-10. Epub 2011 Feb 18. Eukaryot Cell. 2011. PMID: 21335529 Free PMC article.
-
The role of phenotypic switching in the basic biology and pathogenesis of Candida albicans.J Oral Microbiol. 2014 Jan 15;6. doi: 10.3402/jom.v6.22993. eCollection 2014 Jan 15. J Oral Microbiol. 2014. PMID: 24455104 Free PMC article. Review.
-
Fungal biofilms, drug resistance, and recurrent infection.Cold Spring Harb Perspect Med. 2014 Oct 1;4(10):a019729. doi: 10.1101/cshperspect.a019729. Cold Spring Harb Perspect Med. 2014. PMID: 25274758 Free PMC article. Review.
-
Assessment and Optimizations of Candida albicans In Vitro Biofilm Assays.Antimicrob Agents Chemother. 2017 Apr 24;61(5):e02749-16. doi: 10.1128/AAC.02749-16. Print 2017 May. Antimicrob Agents Chemother. 2017. PMID: 28289028 Free PMC article.
References
-
- Almirante, B., D. Rodriguez, M. Cuenca-Estrella, M. Almela, F. Sanchez, J. Ayats, C. Alonso-Tarres, J. L. Rodriguez-Tudela, and A. Pahissa. 2006. Epidemiology, risk factors, and prognosis of Candida parapsilosis bloodstream infections: case-control population-based surveillance study of patients in Barcelona, Spain, from 2002 to 2003. J. Clin. Microbiol. 44:1681-1685. - PMC - PubMed
-
- Bain, J. M., C. Stubberfield, and N. A. Gow. 2001. Ura-status-dependent adhesion of Candida albicans mutants. FEMS Microbiol. Lett. 204:323-328. - PubMed
-
- Bakir, M., N. Cerikcioglu, R. Barton, and A. Yagci. 2006. Epidemiology of candidemia in a Turkish tertiary care hospital. APMIS 114:601-610. - PubMed
-
- Barchiesi, F., G. Caggiano, L. Falconi Di Francesco, M. T. Montagna, S. Barbuti, and G. Scalise. 2004. Outbreak of fungemia due to Candida parapsilosis in a pediatric oncology unit. Diagn. Microbiol. Infect. Dis. 49:269-271. - PubMed
-
- Blankenship, J. R., and A. P. Mitchell. 2006. How to build a biofilm: a fungal perspective. Curr. Opin. Microbiol. 9:588-594. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
