

SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome
- PMID: 17592081
- PMCID: PMC3332546
- DOI: 10.1161/CIRCULATIONAHA.106.659086
SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome
Abstract
Background: Congenital long-QT syndrome (LQTS) is potentially lethal secondary to malignant ventricular arrhythmias and is caused predominantly by mutations in genes that encode cardiac ion channels. Nearly 25% of patients remain without a genetic diagnosis, and genes that encode cardiac channel regulatory proteins represent attractive candidates. Voltage-gated sodium channels have a pore-forming alpha-subunit associated with 1 or more auxiliary beta-subunits. Four different beta-subunits have been described. All are detectable in cardiac tissue, but none have yet been linked to any heritable arrhythmia syndrome.
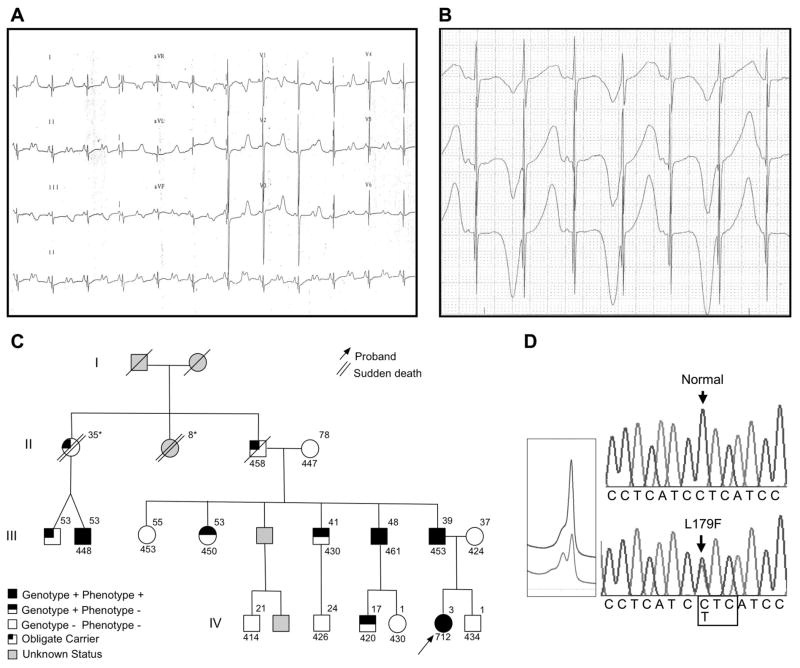
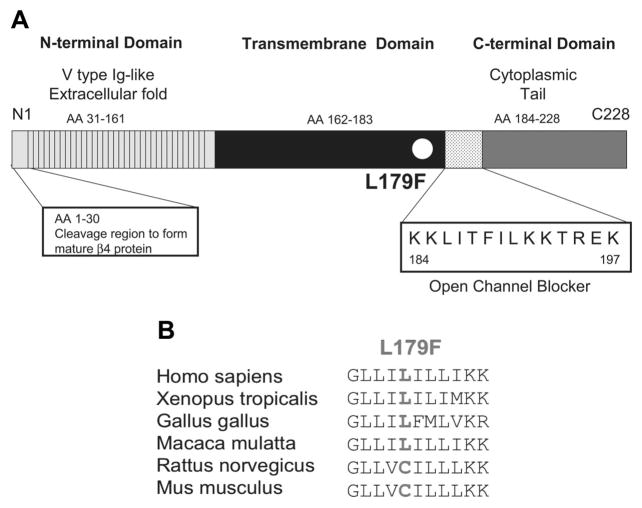
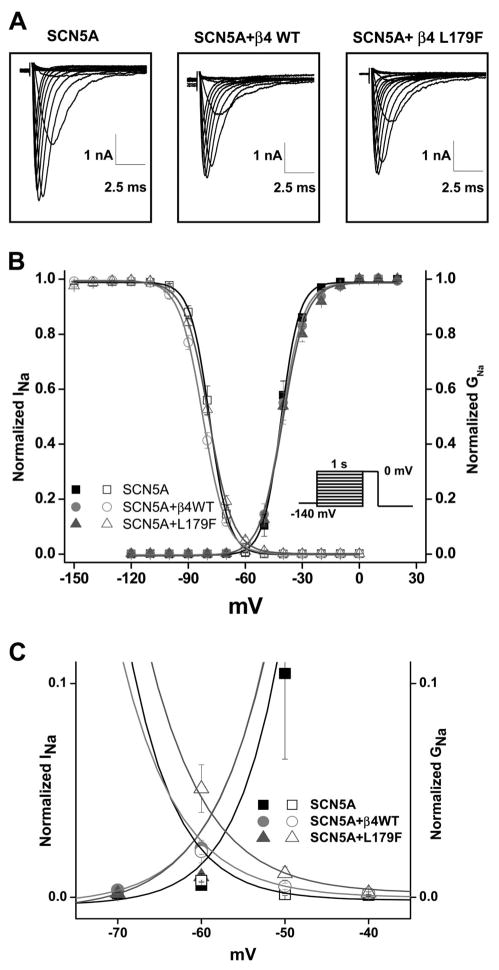
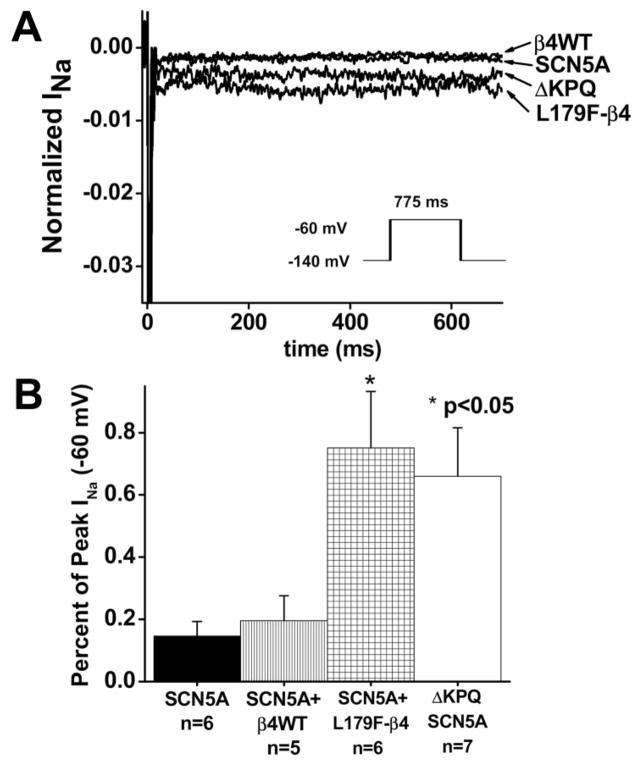
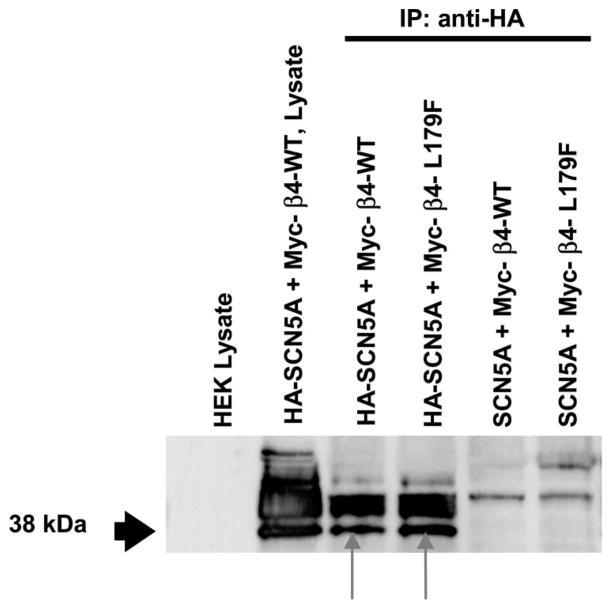
Methods and results: We present a case of a 21-month-old Mexican-mestizo female with intermittent 2:1 atrioventricular block and a corrected QT interval of 712 ms. Comprehensive open reading frame/splice mutational analysis of the 9 established LQTS-susceptibility genes proved negative, and complete mutational analysis of the 4 Na(vbeta)-subunits revealed a L179F (C535T) missense mutation in SCN4B that cosegregated properly throughout a 3-generation pedigree and was absent in 800 reference alleles. After this discovery, SCN4B was analyzed in 262 genotype-negative LQTS patients (96% white), but no further mutations were found. L179F was engineered by site-directed mutagenesis and heterologously expressed in HEK293 cells that contained the stably expressed SCN5A-encoded sodium channel alpha-subunit (hNa(V)1.5). Compared with the wild-type, L179F-beta4 caused an 8-fold (compared with SCN5A alone) and 3-fold (compared with SCN5A + WT-beta4) increase in late sodium current consistent with the molecular/electrophysiological phenotype previously shown for LQTS-associated mutations.
Conclusions: We provide the seminal report of SCN4B-encoded Na(vbeta)4 as a novel LQT3-susceptibility gene.
Figures
References
-
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. - PubMed
-
- Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–11. - PubMed
-
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. - PubMed
-
- Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice [see comment] JAMA. 2005;294:2975–2980. - PubMed
-
- Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67:448–458. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
