

Human population structure detection via multilocus genotype clustering
- PMID: 17592628
- PMCID: PMC1934381
- DOI: 10.1186/1471-2156-8-34
Human population structure detection via multilocus genotype clustering
Abstract
Background: We describe a hierarchical clustering algorithm for using Single Nucleotide Polymorphism (SNP) genetic data to assign individuals to populations. The method does not assume Hardy-Weinberg equilibrium and linkage equilibrium among loci in sample population individuals.
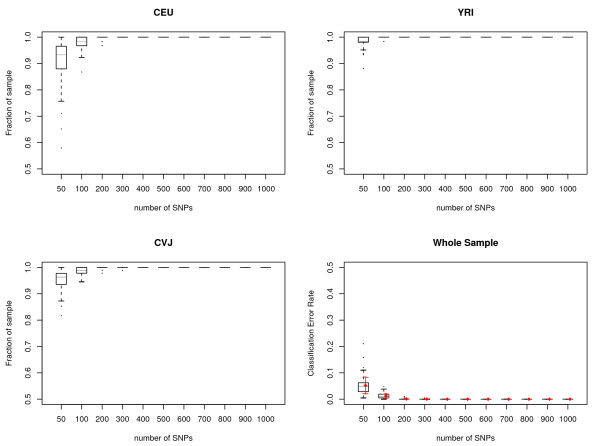
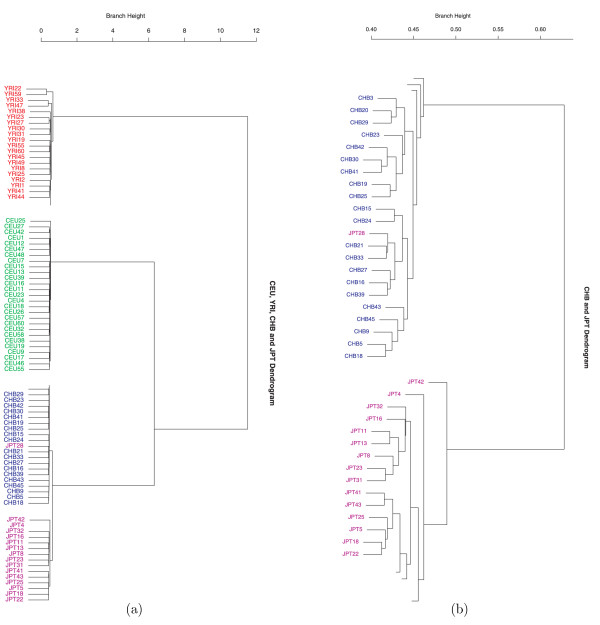
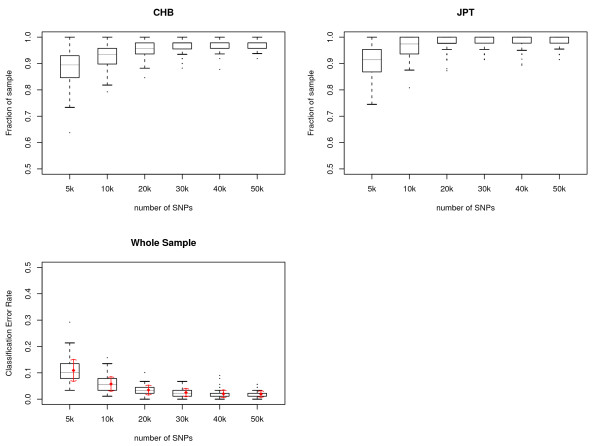
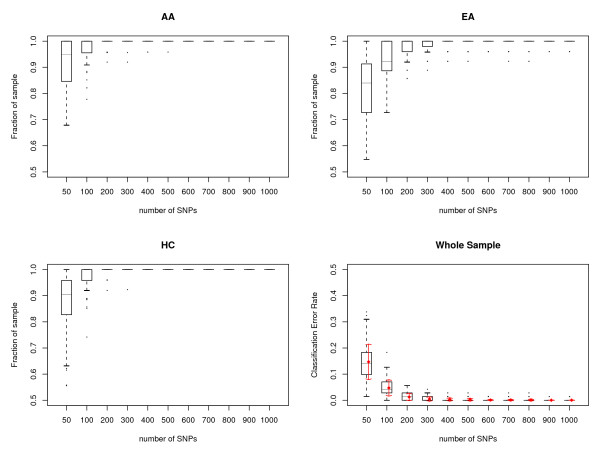
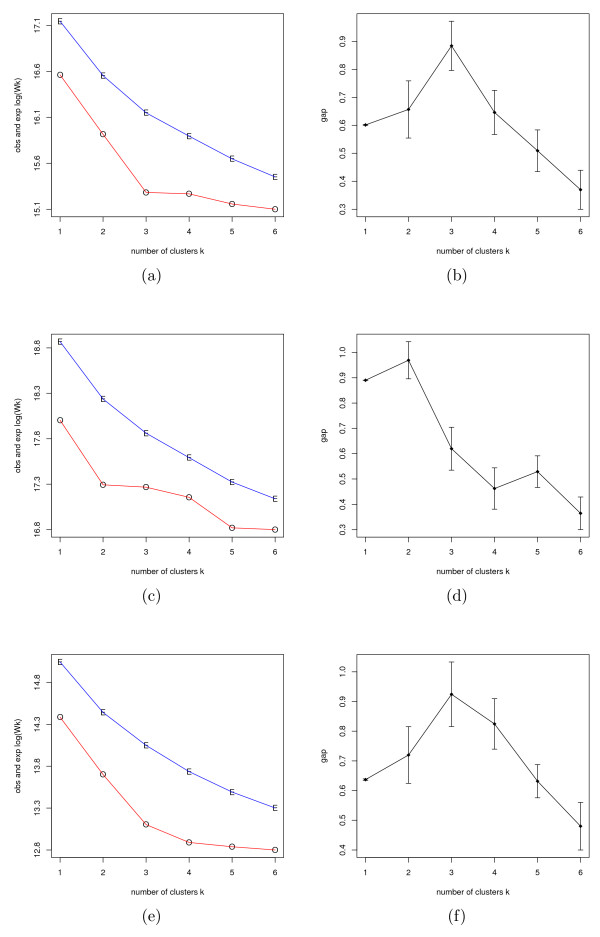
Results: We show that the algorithm can assign sample individuals highly accurately to their corresponding ethnic groups in our tests using HapMap SNP data and it is also robust to admixed populations when tested with Perlegen SNP data. Moreover, it can detect fine-scale population structure as subtle as that between Chinese and Japanese by using genome-wide high-diversity SNP loci.
Conclusion: The algorithm provides an alternative approach to the popular STRUCTURE program, especially for fine-scale population structure detection in genome-wide association studies. This is the first successful separation of Chinese and Japanese samples using random SNP loci with high statistical support.
Figures


References
-
- Freedman M, Reich D, Penney K, McDonald G, Mignault A, Patterson N, Gabriel S, Topol E, Smoller J, Pato C, Pato M, Petryshen T, Kolonel L, Lander E, Sklar P, Henderson B, Hirschhorn J, Altshuler D. Assessing the impact of population stratification on genetic association studies. Nat Genet. 2004;36:388–393. doi: 10.1038/ng1333. - DOI - PubMed
-
- Cavalli-Sforza LL, Menozzi P, Piazza A. In: The history and geography of human genes. Princeton, NJ, editor. Princeton University Press; 1994.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
