

High-throughput identification of interacting protein-protein binding sites
- PMID: 17594507
- PMCID: PMC1925121
- DOI: 10.1186/1471-2105-8-223
High-throughput identification of interacting protein-protein binding sites
Abstract
Background: With the advent of increasing sequence and structural data, a number of methods have been proposed to locate putative protein binding sites from protein surfaces. Therefore, methods that are able to identify whether these binding sites interact are needed.
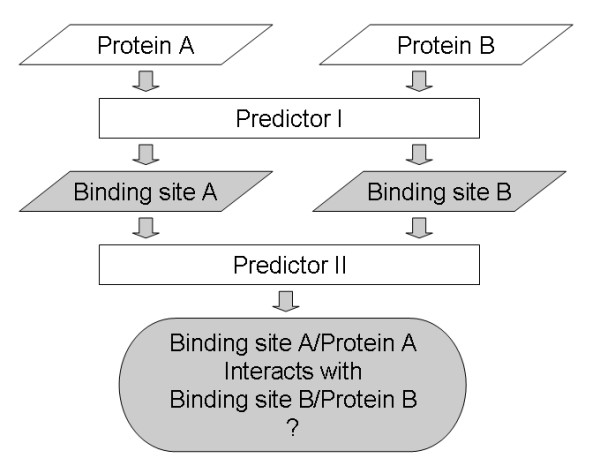
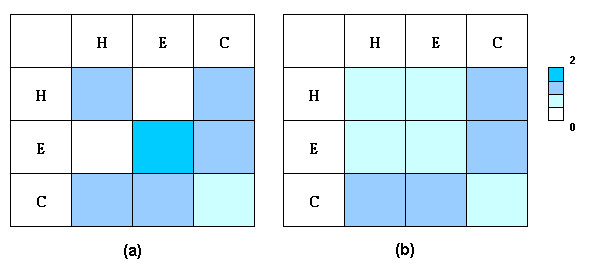
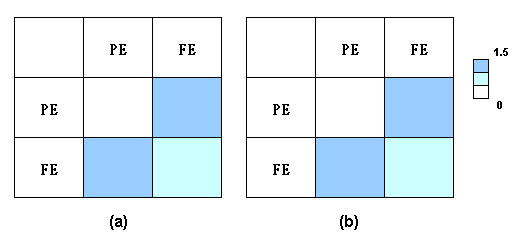
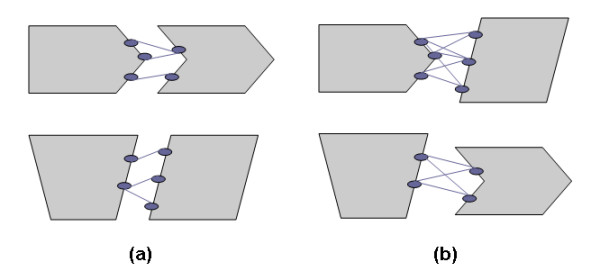
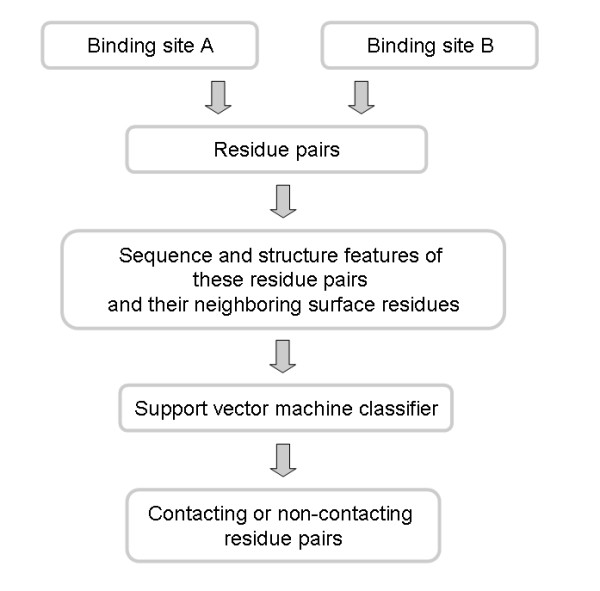
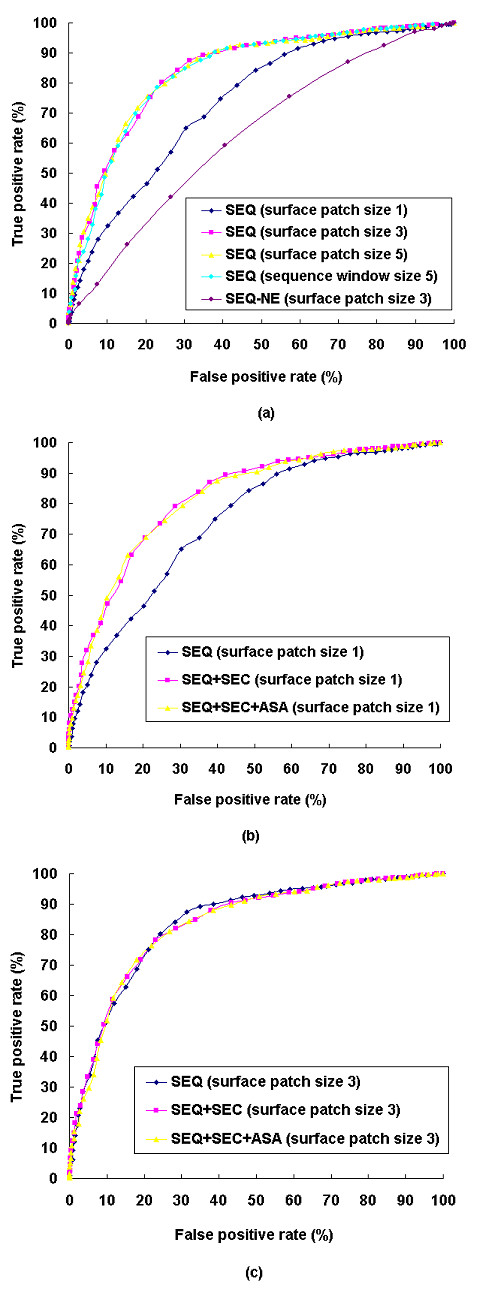
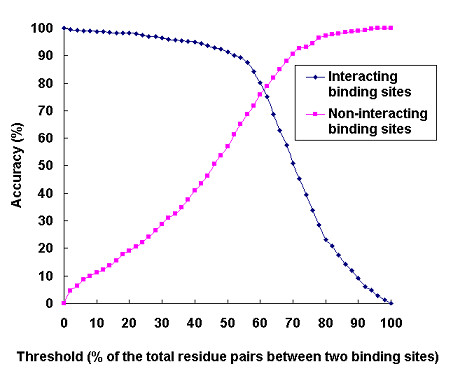
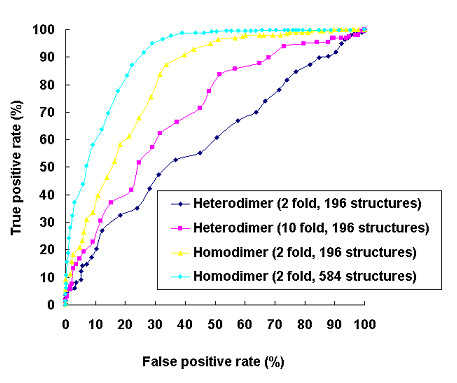
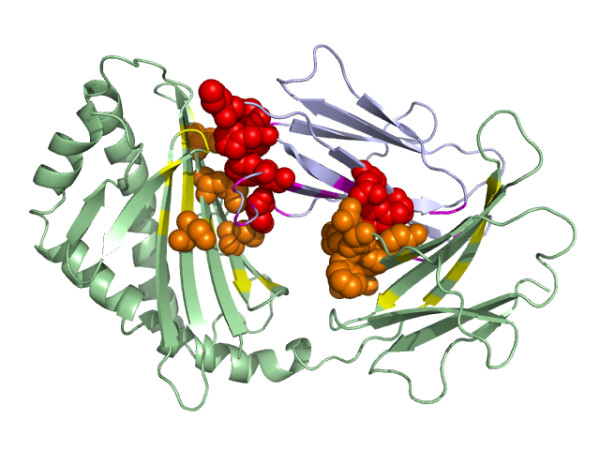
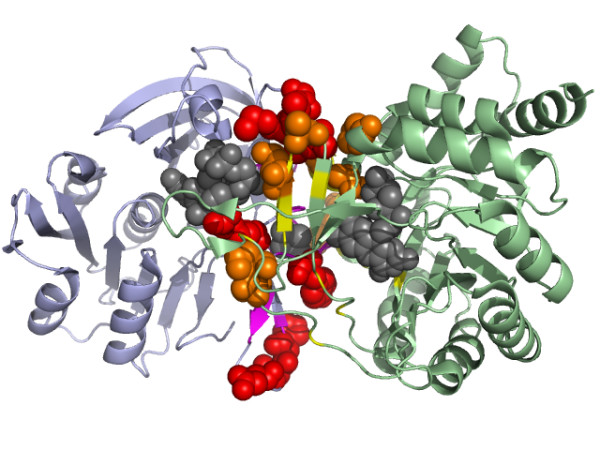
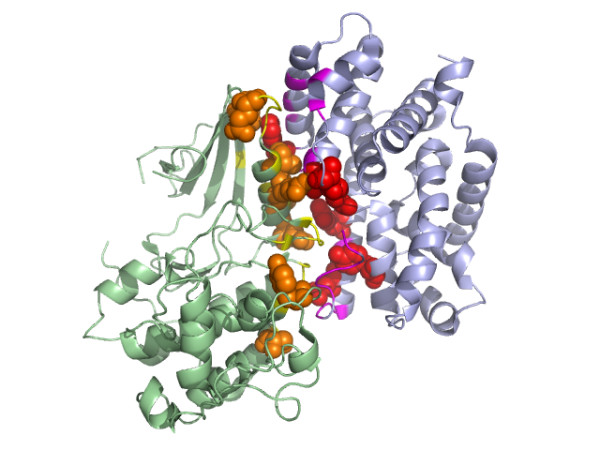
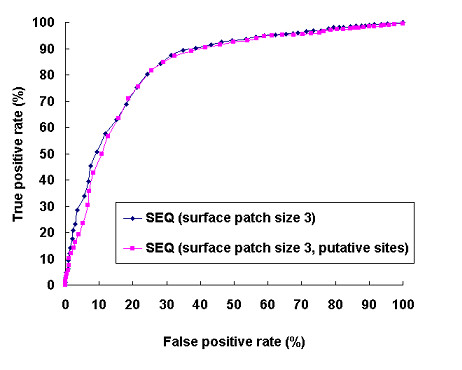
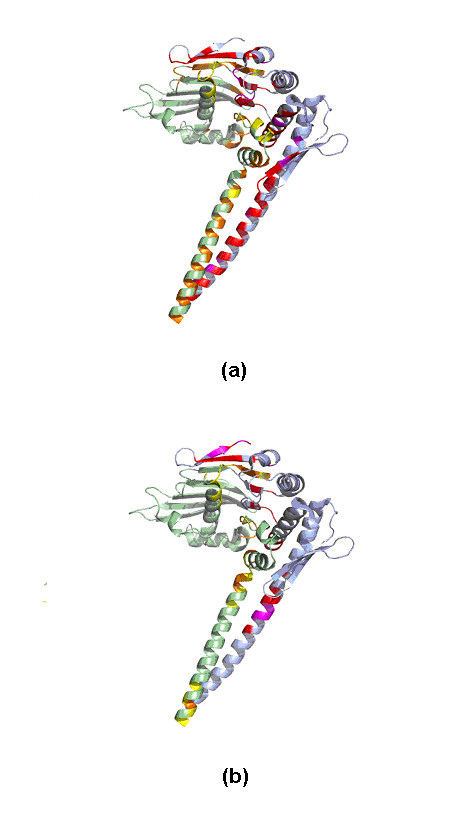
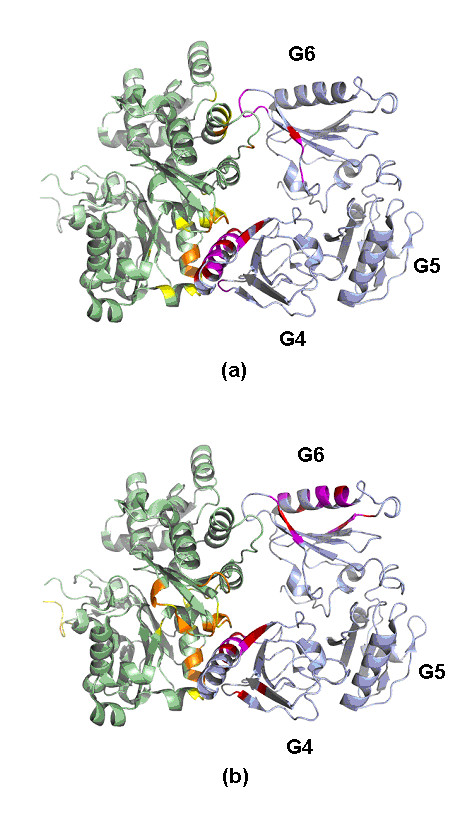
Results: We have developed a new method using a machine learning approach to detect if protein binding sites, once identified, interact with each other. The method exploits information relating to sequence and structural complementary across protein interfaces and has been tested on a non-redundant data set consisting of 584 homo-dimers and 198 hetero-dimers extracted from the PDB. Results indicate 87.4% of the interacting binding sites and 68.6% non-interacting binding sites were correctly identified. Furthermore, we built a pipeline that links this method to a modified version of our previously developed method that predicts the location of binding sites.
Conclusion: We have demonstrated that this high-throughput pipeline is capable of identifying binding sites for proteins, their interacting binding sites and, ultimately, their binding partners on a large scale.
Figures

Similar articles
-
ISIS: interaction sites identified from sequence.Bioinformatics. 2007 Jan 15;23(2):e13-6. doi: 10.1093/bioinformatics/btl303. Bioinformatics. 2007. PMID: 17237081
-
Moment invariants as shape recognition technique for comparing protein binding sites.Bioinformatics. 2007 Dec 1;23(23):3139-46. doi: 10.1093/bioinformatics/btm503. Epub 2007 Oct 31. Bioinformatics. 2007. PMID: 17977888
-
Modelling interaction sites in protein domains with interaction profile hidden Markov models.Bioinformatics. 2006 Dec 1;22(23):2851-7. doi: 10.1093/bioinformatics/btl486. Epub 2006 Sep 25. Bioinformatics. 2006. PMID: 17000753
-
Analyzing molecular interactions.Curr Protoc Bioinformatics. 2003 May;Chapter 8:Unit8.1. doi: 10.1002/0471250953.bi0801s01. Curr Protoc Bioinformatics. 2003. PMID: 18428708 Review.
-
Interaction-site prediction for protein complexes: a critical assessment.Bioinformatics. 2007 Sep 1;23(17):2203-9. doi: 10.1093/bioinformatics/btm323. Epub 2007 Jun 22. Bioinformatics. 2007. PMID: 17586545 Review.
Cited by
-
DockRank: ranking docked conformations using partner-specific sequence homology-based protein interface prediction.Proteins. 2014 Feb;82(2):250-67. doi: 10.1002/prot.24370. Epub 2013 Oct 17. Proteins. 2014. PMID: 23873600 Free PMC article.
-
Ranking Docked Models of Protein-Protein Complexes Using Predicted Partner-Specific Protein-Protein Interfaces: A Preliminary Study.ACM BCB. 2011 Aug;2011:441-445. doi: 10.1145/2147805.2147866. ACM BCB. 2011. PMID: 25905110 Free PMC article.
-
Prediction of Protein-Protein Interaction Sites Based on Stratified Attentional Mechanisms.Front Genet. 2021 Nov 22;12:784863. doi: 10.3389/fgene.2021.784863. eCollection 2021. Front Genet. 2021. PMID: 34880910 Free PMC article.
-
Multi-level learning: improving the prediction of protein, domain and residue interactions by allowing information flow between levels.BMC Bioinformatics. 2009 Aug 5;10:241. doi: 10.1186/1471-2105-10-241. BMC Bioinformatics. 2009. PMID: 19656385 Free PMC article.
-
Predicting protein-protein interface residues using local surface structural similarity.BMC Bioinformatics. 2012 Mar 18;13:41. doi: 10.1186/1471-2105-13-41. BMC Bioinformatics. 2012. PMID: 22424103 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
