

Predicting absolute ligand binding free energies to a simple model site
- PMID: 17599350
- PMCID: PMC2104542
- DOI: 10.1016/j.jmb.2007.06.002
Predicting absolute ligand binding free energies to a simple model site
Abstract
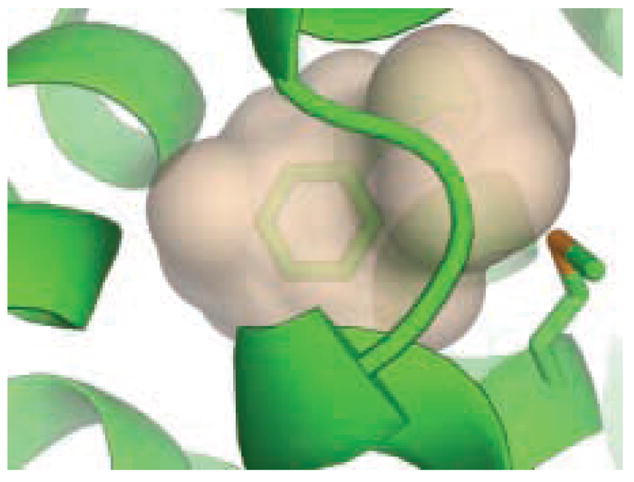
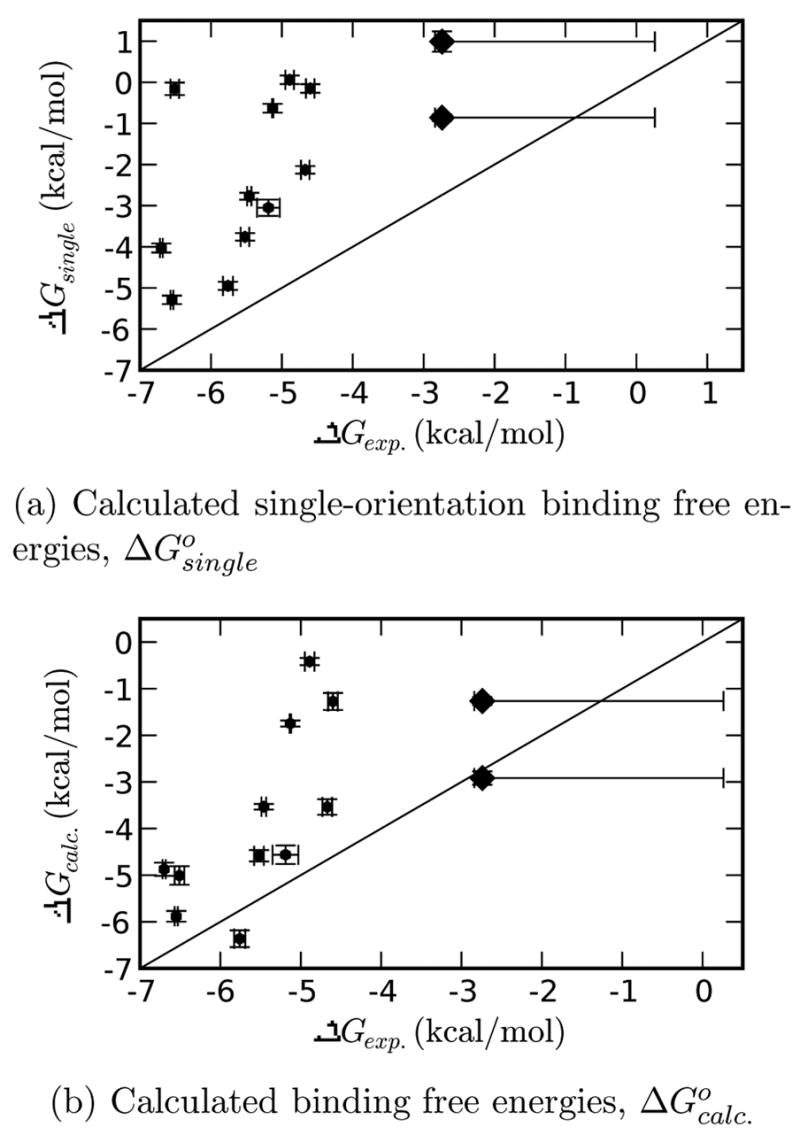
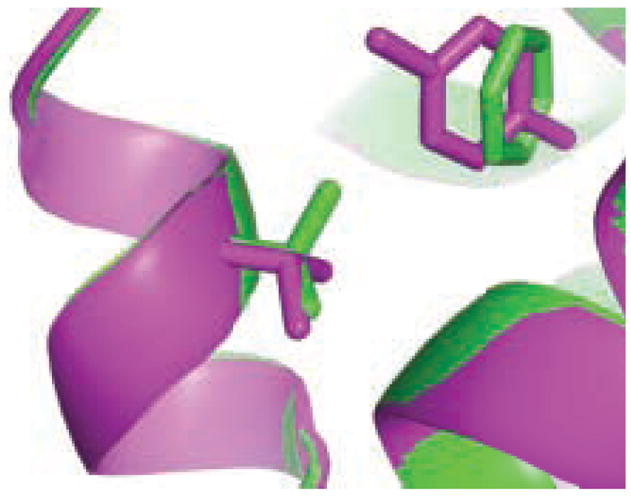
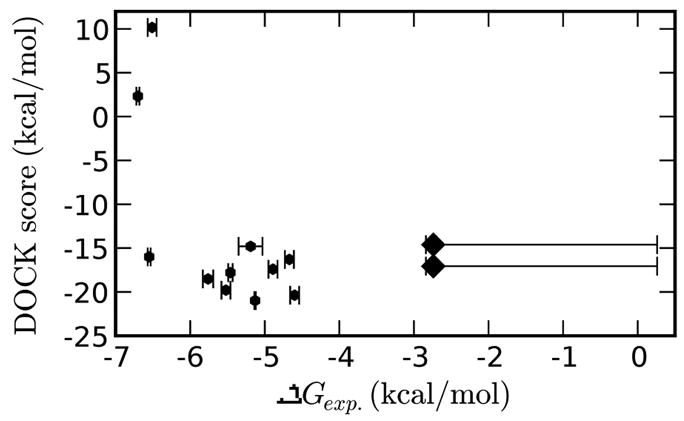
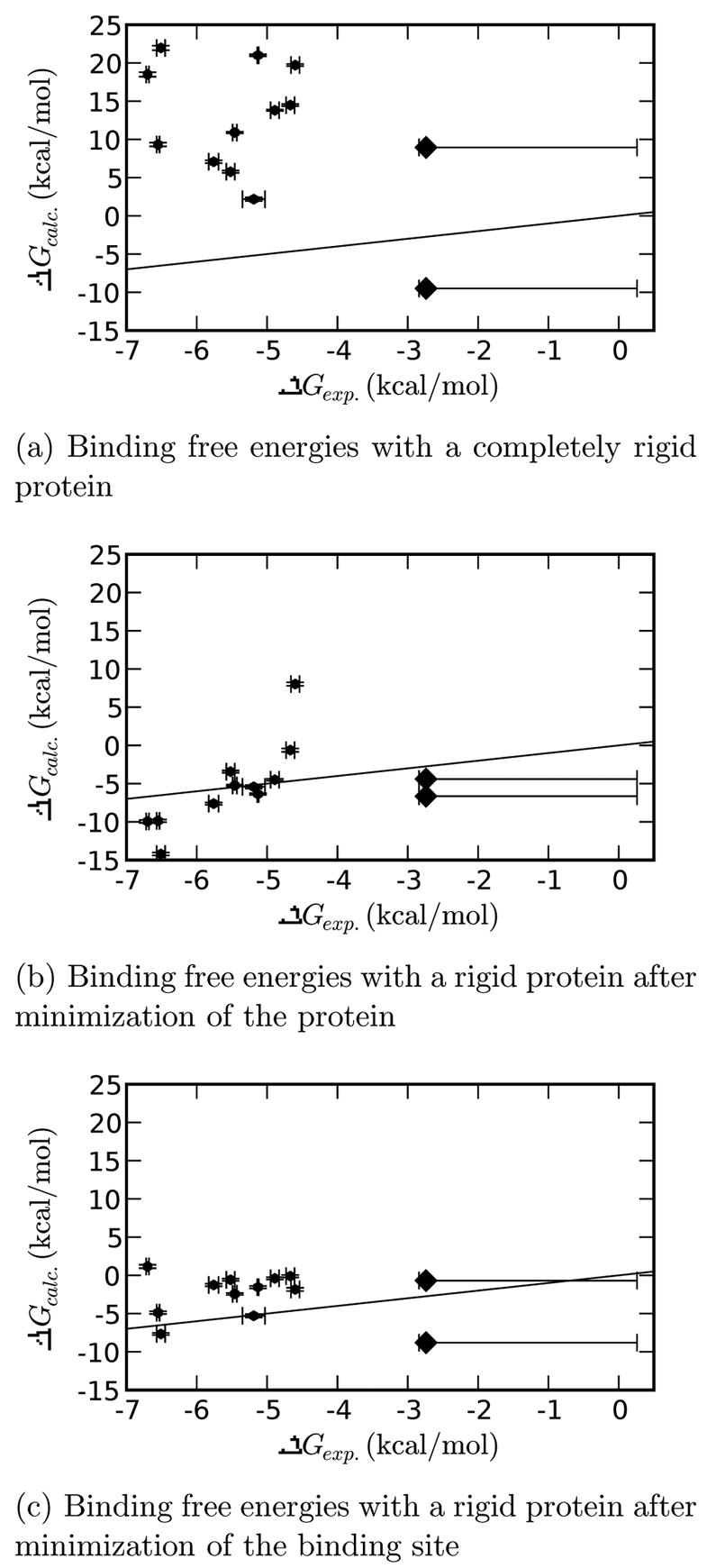
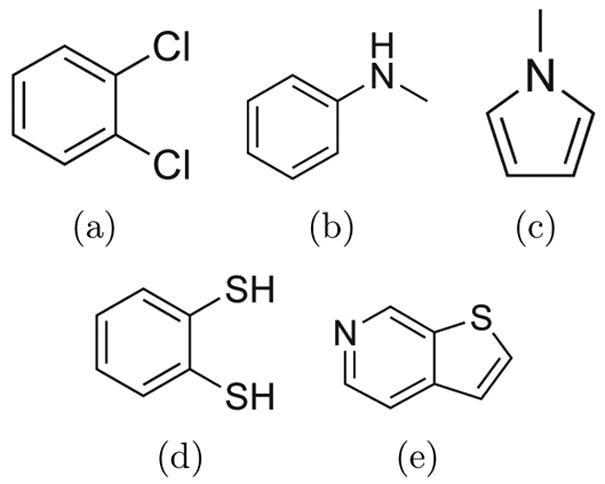
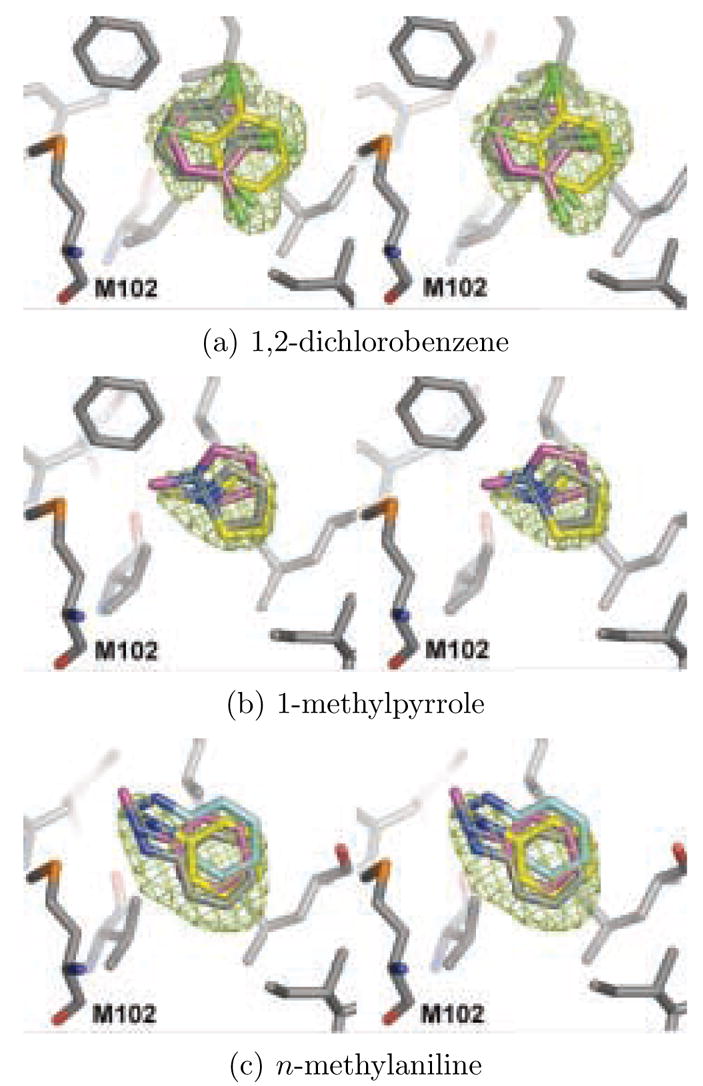
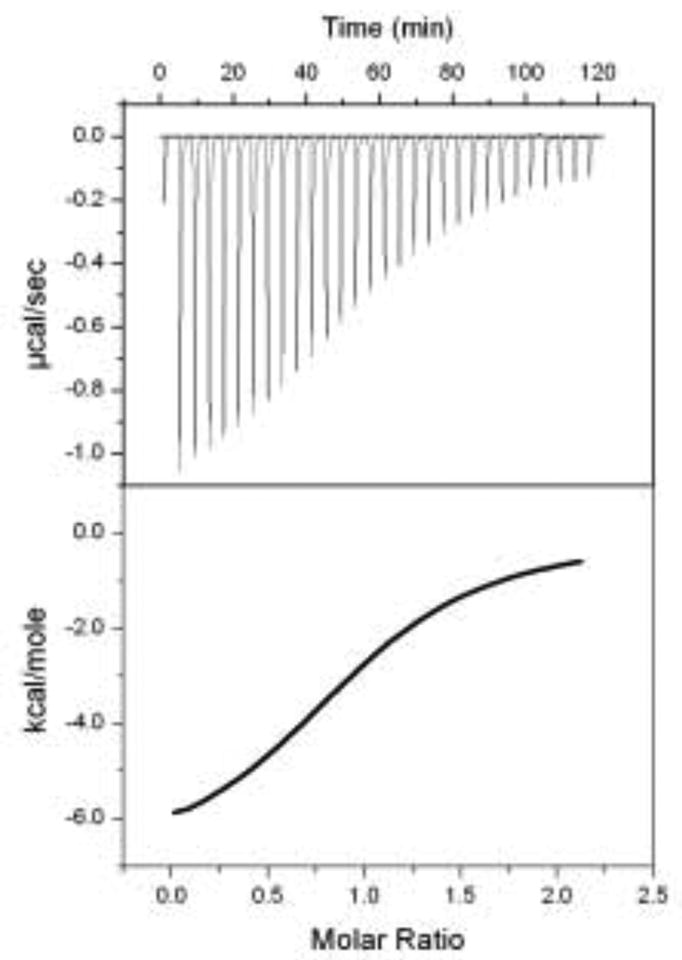
A central challenge in structure-based ligand design is the accurate prediction of binding free energies. Here we apply alchemical free energy calculations in explicit solvent to predict ligand binding in a model cavity in T4 lysozyme. Even in this simple site, there are challenges. We made systematic improvements, beginning with single poses from docking, then including multiple poses, additional protein conformational changes, and using an improved charge model. Computed absolute binding free energies had an RMS error of 1.9 kcal/mol relative to previously determined experimental values. In blind prospective tests, the methods correctly discriminated between several true ligands and decoys in a set of putative binders identified by docking. In these prospective tests, the RMS error in predicted binding free energies relative to those subsequently determined experimentally was only 0.6 kcal/mol. X-ray crystal structures of the new ligands bound in the cavity corresponded closely to predictions from the free energy calculations, but sometimes differed from those predicted by docking. Finally, we examined the impact of holding the protein rigid, as in docking, with a view to learning how approximations made in docking affect accuracy and how they may be improved.
Figures
References
-
- Halperin I, Ma B, Wolfson H, Nussinov R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins: Str, Funct, and Genet. 2002;47:409–443. - PubMed
-
- Shoichet BK, Leach AR, Kuntz ID. Ligand solvation in molecular docking. Proteins: Struct, Funct, and Genet. 1999;34:4–16. - PubMed
-
- Huang N, Kalyanaraman C, Bernacki K, Jacobson MP. Molecular mechanics methods for predicting protein-ligand binding. Phys Chem Chem Phys. 2006;8:5166–5177. - PubMed
-
- Srinivasan J, Cheatam TE, III, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA, and Phosphoramidate-DNA helices. J Am Chem Soc. 1998;120:9401–9409.
-
- Kuhn B, Kollman PA. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J Med Chem. 2000;43:3786–3791. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
