

TGF-beta1 stimulates human AT1 receptor expression in lung fibroblasts by cross talk between the Smad, p38 MAPK, JNK, and PI3K signaling pathways
- PMID: 17601799
- PMCID: PMC2413071
- DOI: 10.1152/ajplung.00099.2007
TGF-beta1 stimulates human AT1 receptor expression in lung fibroblasts by cross talk between the Smad, p38 MAPK, JNK, and PI3K signaling pathways
Retraction in
-
Retraction.Am J Physiol Lung Cell Mol Physiol. 2012 Apr 1;302(7):L719. doi: 10.1152/ajplung.zh5-6097-retr.2012. Am J Physiol Lung Cell Mol Physiol. 2012. PMID: 22467678 Free PMC article. No abstract available.
Abstract
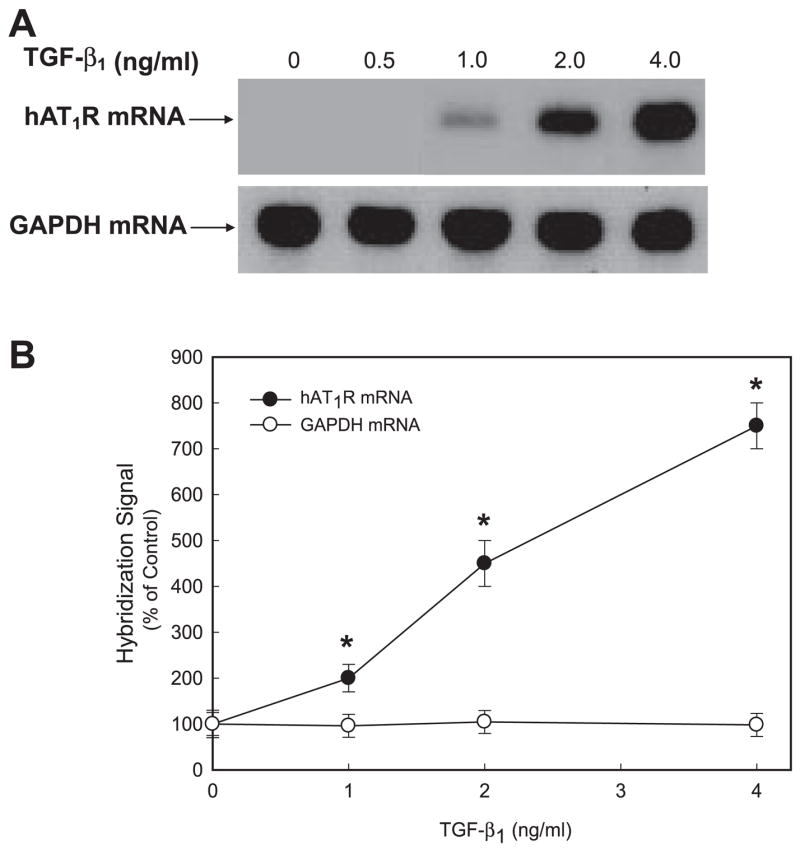
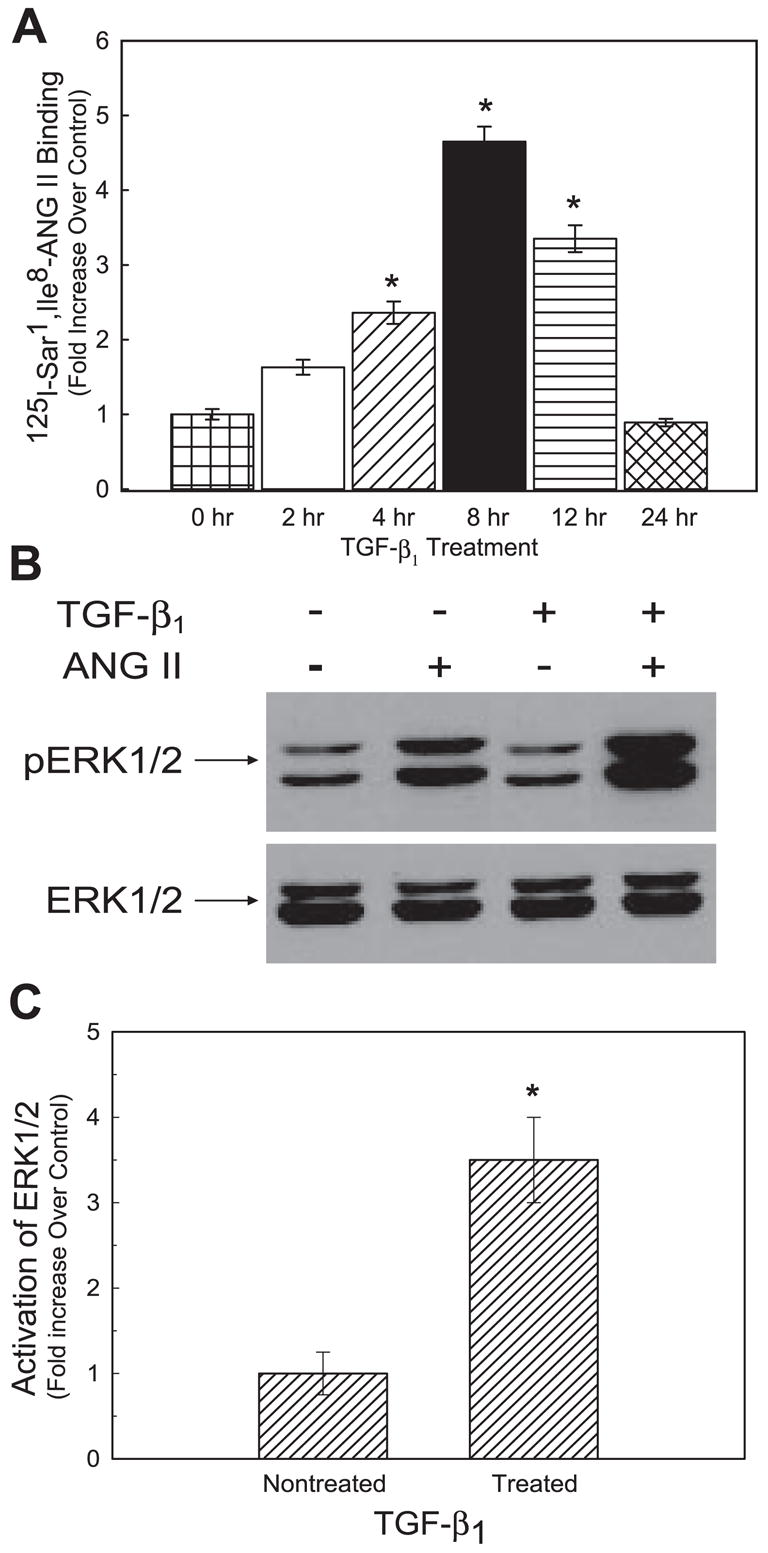
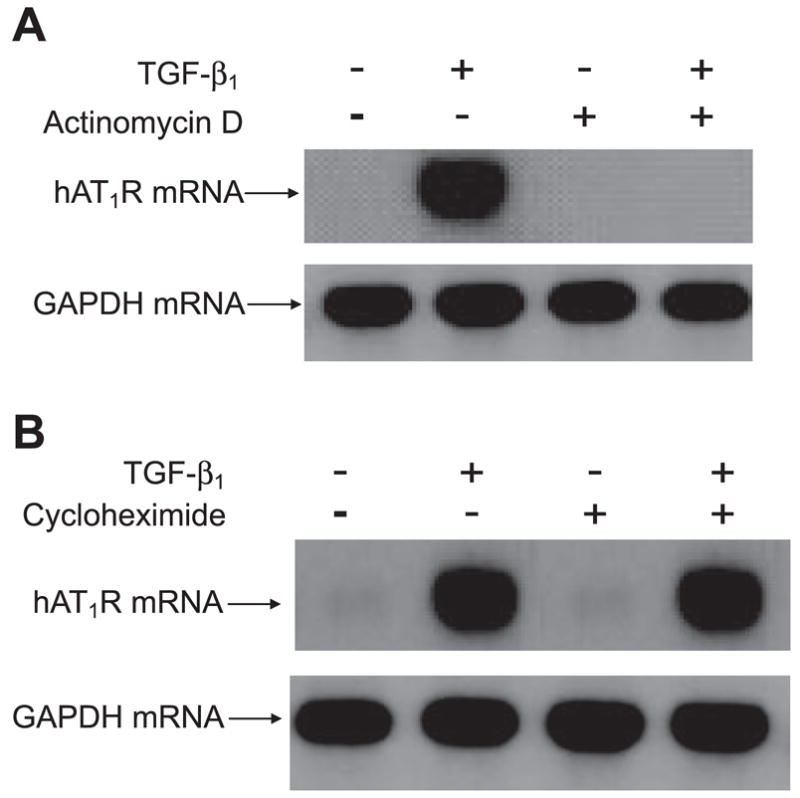
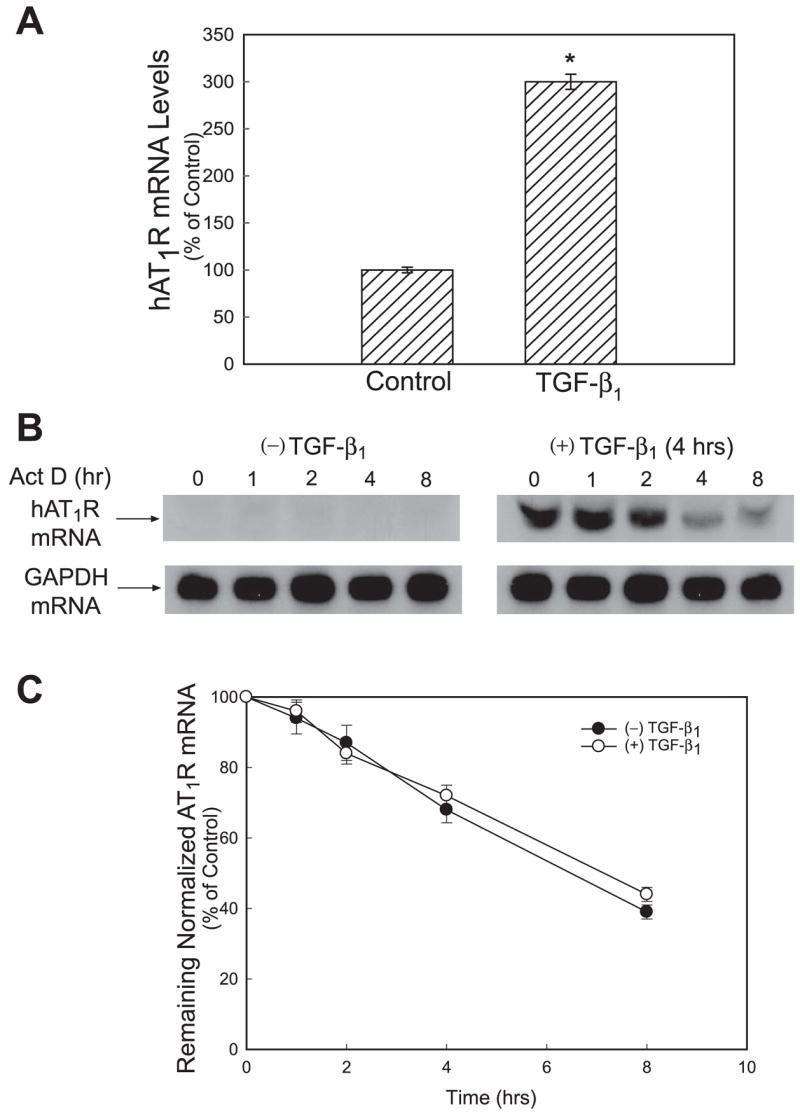
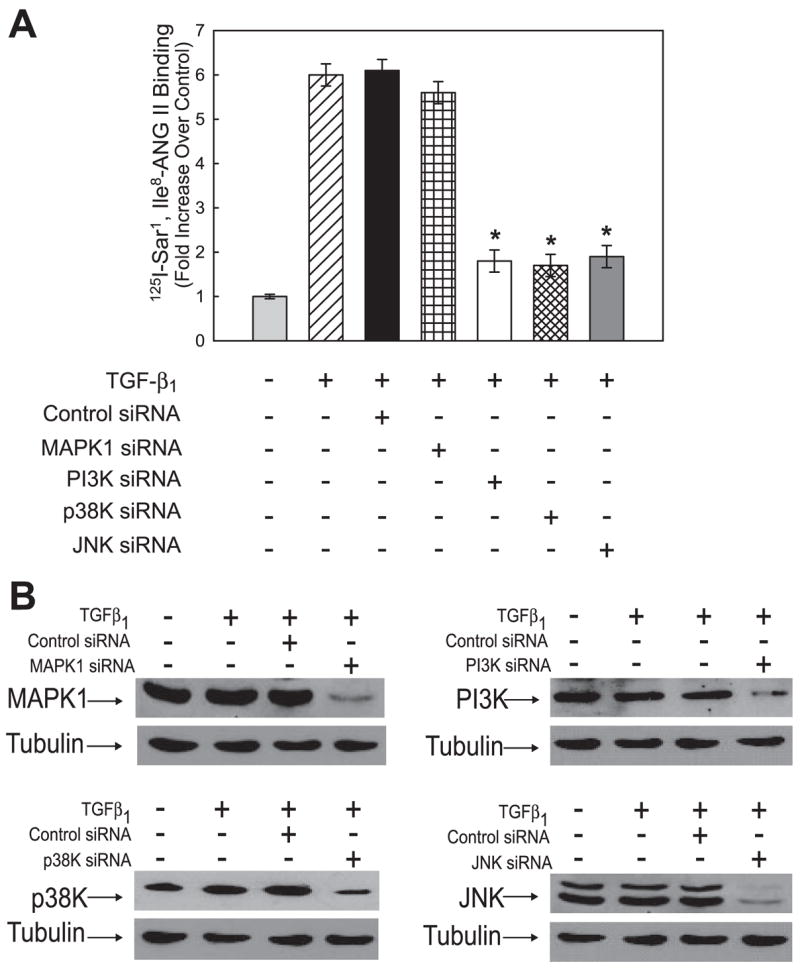
Both angiotensin II (ANG II) and transforming growth factor-beta1 (TGF-beta1) are thought to be involved in mediating pulmonary fibrosis. Interactions between the renin-angiotensin system (RAS) and TGF-beta1 have been well documented, with most studies describing the effect of ANG II on TGF-beta1 expression. However, recent gene expression profiling experiments demonstrated that the angiotensin II type 1 receptor (AT(1)R) gene was a novel TGF-beta1 target in human adult lung fibroblasts. In this report, we show that TGF-beta1 augments human AT(1)R (hAT(1)R) steady-state mRNA and protein levels in a dose- and time-dependent manner in primary human fetal pulmonary fibroblasts (hPFBs). Nuclear run-on experiments demonstrate that TGF-beta1 transcriptionally activates the hAT(1)R gene and does not influence hAT(1)R mRNA stability. Pharmacological inhibitors and specific siRNA knockdown experiments demonstrate that the TGF-beta1 type 1 receptor (TbetaRI/ALK5), Smad2/3, and Smad4 are essential for TGF-beta1-stimulated hAT(1)R expression. Additional pharmacological inhibitor and small interference RNA experiments also demonstrated that p38 MAPK, JNK, and phosphatidylinositol 3-kinase (PI3K) signaling pathways are also involved in the TGF-beta1-stimulated increase in hAT(1)R density. Together, our results suggest an important role for cross talk among Smad, p38 MAPK, JNK, and PI3K pathways in mediating the augmented expression of hAT(1)R following TGF-beta1 treatment in hPFB. This study supports the hypothesis that a self-potentiating loop exists between the RAS and the TGF-beta1 signaling pathways and suggests that ANG II and TGF-beta1 may cooperate in the pathogenesis of pulmonary fibrosis. The synergy between these systems may require that both pathways be simultaneously inhibited to treat fibrotic lung disease.
Figures





Comment in
-
Findings of research misconduct.NIH Guide Grants Contracts (Bethesda). 2013 Jan 18:NOT-OD-13-023. NIH Guide Grants Contracts (Bethesda). 2013. PMID: 23367540 Free PMC article. No abstract available.
Similar articles
-
C-Jun-NH2-terminal kinase mediates expression of connective tissue growth factor induced by transforming growth factor-beta1 in human lung fibroblasts.Am J Respir Cell Mol Biol. 2003 Jun;28(6):754-61. doi: 10.1165/rcmb.4892. Am J Respir Cell Mol Biol. 2003. PMID: 12760970
-
Tissue-specific mechanisms for CCN2/CTGF persistence in fibrotic gingiva: interactions between cAMP and MAPK signaling pathways, and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase.J Biol Chem. 2007 May 25;282(21):15416-29. doi: 10.1074/jbc.M610432200. Epub 2007 Apr 10. J Biol Chem. 2007. PMID: 17428796 Free PMC article.
-
Endothelin-1 and transforming growth factor-beta1 independently induce fibroblast resistance to apoptosis via AKT activation.Am J Respir Cell Mol Biol. 2009 Oct;41(4):484-93. doi: 10.1165/rcmb.2008-0447OC. Epub 2009 Feb 2. Am J Respir Cell Mol Biol. 2009. PMID: 19188658 Free PMC article.
-
Glycosyltransferases and glycosaminoglycans in bleomycin and transforming growth factor-β1-induced pulmonary fibrosis.Am J Respir Cell Mol Biol. 2014 Mar;50(3):583-94. doi: 10.1165/rcmb.2012-0226OC. Am J Respir Cell Mol Biol. 2014. PMID: 24127863
-
Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling.Cytokine. 2011 Jul;55(1):90-7. doi: 10.1016/j.cyto.2011.03.024. Epub 2011 Apr 17. Cytokine. 2011. PMID: 21498085
Cited by
-
The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-β-Induced Epithelial-Mesenchymal Transition.J Signal Transduct. 2012;2012:289243. doi: 10.1155/2012/289243. Epub 2012 Jan 29. J Signal Transduct. 2012. PMID: 22363839 Free PMC article.
-
A new antifibrotic target of Ac-SDKP: inhibition of myofibroblast differentiation in rat lung with silicosis.PLoS One. 2012;7(7):e40301. doi: 10.1371/journal.pone.0040301. Epub 2012 Jul 3. PLoS One. 2012. PMID: 22802960 Free PMC article.
-
Selective p38α mitogen-activated protein kinase inhibitor attenuates lung inflammation and fibrosis in IL-13 transgenic mouse model of asthma.J Asthma Allergy. 2008 Nov 16;1:31-44. doi: 10.2147/jaa.s4199. J Asthma Allergy. 2008. PMID: 21436983 Free PMC article.
-
Aldose reductase inhibition prevents allergic airway remodeling through PI3K/AKT/GSK3β pathway in mice.PLoS One. 2013;8(2):e57442. doi: 10.1371/journal.pone.0057442. Epub 2013 Feb 27. PLoS One. 2013. PMID: 23460857 Free PMC article.
-
Mast cells: a pivotal role in pulmonary fibrosis.DNA Cell Biol. 2013 Apr;32(4):206-18. doi: 10.1089/dna.2013.2005. DNA Cell Biol. 2013. PMID: 23570576 Free PMC article.
References
-
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. - PubMed
-
- Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–765. - PubMed
-
- Brezniceanu ML, Wei CC, Zhang SL, Hsieh TJ, Guo DF, Hebert MJ, Ingelfinger JR, Filep JG, Chan JSD. Transforming growth factor-β1 stimulates angiotensinogen gene expression in kidney proximal tubular cells. Kidney Int. 2006;69:1977–1985. - PubMed
-
- Bullock GR, Steyaert I, Bulbe G, Carey RM, Kips J, DePaepe B, Pauwels R, Praet M, Siragy HM, de Gasparo M. Distribution of type-1 and type-2 angiotensin receptors in the normal human lung and in lungs from patients with chronic obstructive pulmonary disease. Histochem Cell Biol. 2001;115:117–124. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous