

Displacement of alpha-actinin from the NMDA receptor NR1 C0 domain By Ca2+/calmodulin promotes CaMKII binding
- PMID: 17602661
- PMCID: PMC2547089
- DOI: 10.1021/bi0623025
Displacement of alpha-actinin from the NMDA receptor NR1 C0 domain By Ca2+/calmodulin promotes CaMKII binding
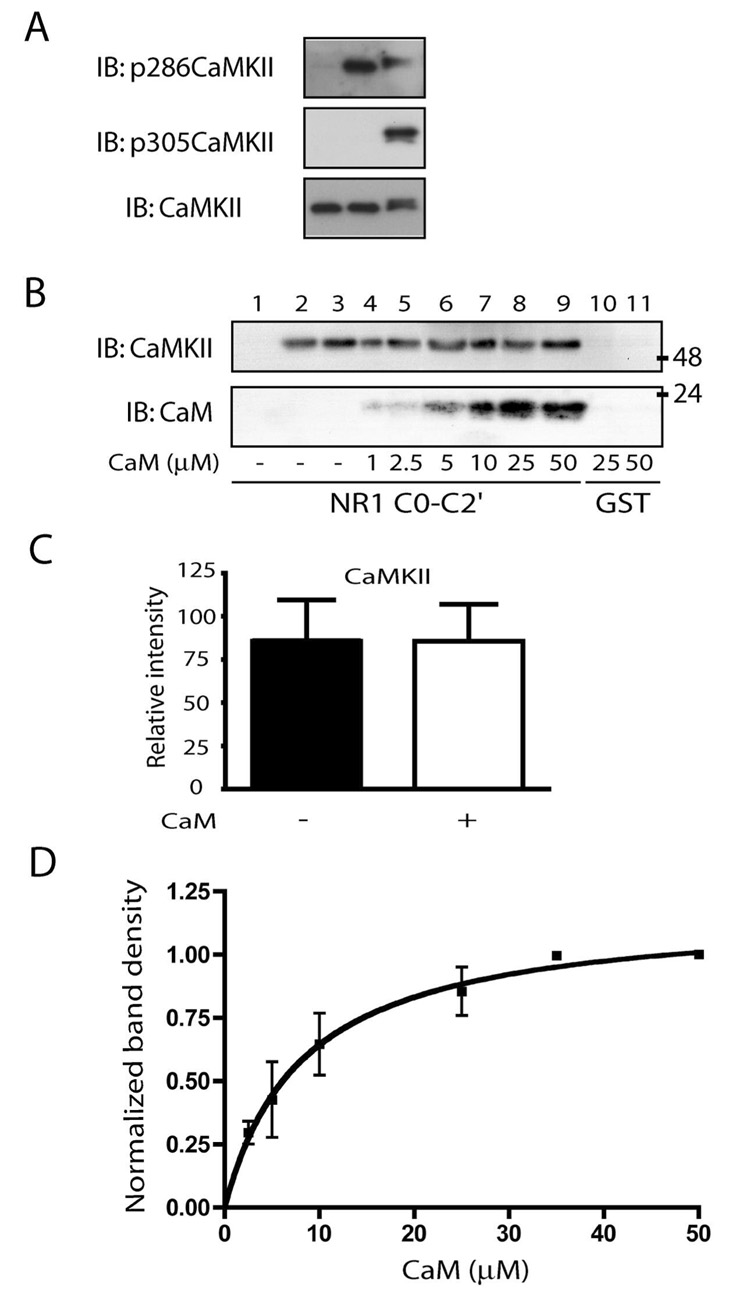
Abstract
Ca2+ influx through the N-methyl-d-aspartate (NMDA)-type glutamate receptor triggers activation and postsynaptic accumulation of Ca2+/calmodulin-dependent kinase II (CaMKII). CaMKII, calmodulin, and alpha-actinin directly bind to the short membrane proximal C0 domain of the C-terminal region of the NMDA receptor NR1 subunit. In a negative feedback loop, calmodulin mediates Ca2+-dependent inactivation of the NMDA receptor by displacing alpha-actinin from NR1 C0 upon Ca2+ influx. We show that Ca2+-depleted calmodulin and alpha-actinin simultaneously bind to NR1 C0. Upon addition of Ca2+, calmodulin dislodges alpha-actinin. Either the N- or C-terminal half of calmodulin is sufficient for Ca2+-induced displacement of alpha-actinin. Whereas alpha-actinin directly antagonizes CaMKII binding to NR1 C0, the addition of Ca2+/calmodulin shifts binding of NR1 C0 toward CaMKII by displacing alpha-actinin. Displacement of alpha-actinin results in the simultaneous binding of calmodulin and CaMKII to NR1 C0. Our results reveal an intricate mechanism whereby Ca2+ functions to govern the complex interactions between the two most prevalent signaling molecules in synaptic plasticity, the NMDA receptor and CaMKII.
Figures





Similar articles
-
Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-D-aspartate receptors by calcium/calmodulin and alpha-actinin.J Biol Chem. 2002 Dec 13;277(50):48441-8. doi: 10.1074/jbc.M205164200. Epub 2002 Oct 13. J Biol Chem. 2002. PMID: 12379661
-
Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-D-aspartate receptor NR2B subunits and alpha-actinin.J Biol Chem. 2005 Nov 25;280(47):39316-23. doi: 10.1074/jbc.M508189200. Epub 2005 Sep 19. J Biol Chem. 2005. PMID: 16172120
-
Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors.J Neurosci. 1999 Feb 15;19(4):1165-78. doi: 10.1523/JNEUROSCI.19-04-01165.1999. J Neurosci. 1999. PMID: 9952395 Free PMC article.
-
Activity-driven postsynaptic translocation of CaMKII.Trends Pharmacol Sci. 2005 Dec;26(12):645-53. doi: 10.1016/j.tips.2005.10.003. Epub 2005 Oct 25. Trends Pharmacol Sci. 2005. PMID: 16253351 Review.
-
Targeting of calcium/calmodulin-dependent protein kinase II.Biochem J. 2004 Feb 15;378(Pt 1):1-16. doi: 10.1042/BJ20031547. Biochem J. 2004. PMID: 14653781 Free PMC article. Review.
Cited by
-
Alternative splice isoforms of small conductance calcium-activated SK2 channels differ in molecular interactions and surface levels.Channels (Austin). 2014;8(1):62-75. doi: 10.4161/chan.27470. Epub 2014 Jan 6. Channels (Austin). 2014. PMID: 24394769 Free PMC article.
-
Actinin-4 Governs Dendritic Spine Dynamics and Promotes Their Remodeling by Metabotropic Glutamate Receptors.J Biol Chem. 2015 Jun 26;290(26):15909-20. doi: 10.1074/jbc.M115.640136. Epub 2015 May 5. J Biol Chem. 2015. PMID: 25944910 Free PMC article.
-
Negative modulation of NMDA receptor channel function by DREAM/calsenilin/KChIP3 provides neuroprotection?Front Mol Neurosci. 2012 Apr 13;5:39. doi: 10.3389/fnmol.2012.00039. eCollection 2012. Front Mol Neurosci. 2012. PMID: 22518099 Free PMC article.
-
Postsynaptic clustering and activation of Pyk2 by PSD-95.J Neurosci. 2010 Jan 13;30(2):449-63. doi: 10.1523/JNEUROSCI.4992-08.2010. J Neurosci. 2010. PMID: 20071509 Free PMC article.
-
Electron cryotomography of postsynaptic densities during development reveals a mechanism of assembly.Neuroscience. 2012 Jun 14;212:19-29. doi: 10.1016/j.neuroscience.2012.03.042. Epub 2012 Apr 16. Neuroscience. 2012. PMID: 22516021 Free PMC article.
References
-
- Soderling TR, Chang B, Brickey D. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. Journal of Biological Chemistry. 2001;276:3719–3722. - PubMed
-
- Malenka RC, Nicoll RA. Long-term potentiation-a decade of progress? Science. 1999;285:1870–1874. - PubMed
-
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. - PubMed
-
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. - PubMed
-
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
