

Towards the high-resolution protein structure prediction. Fast refinement of reduced models with all-atom force field
- PMID: 17603876
- PMCID: PMC1933428
- DOI: 10.1186/1472-6807-7-43
Towards the high-resolution protein structure prediction. Fast refinement of reduced models with all-atom force field
Abstract
Background: Although experimental methods for determining protein structure are providing high resolution structures, they cannot keep the pace at which amino acid sequences are resolved on the scale of entire genomes. For a considerable fraction of proteins whose structures will not be determined experimentally, computational methods can provide valuable information. The value of structural models in biological research depends critically on their quality. Development of high-accuracy computational methods that reliably generate near-experimental quality structural models is an important, unsolved problem in the protein structure modeling.
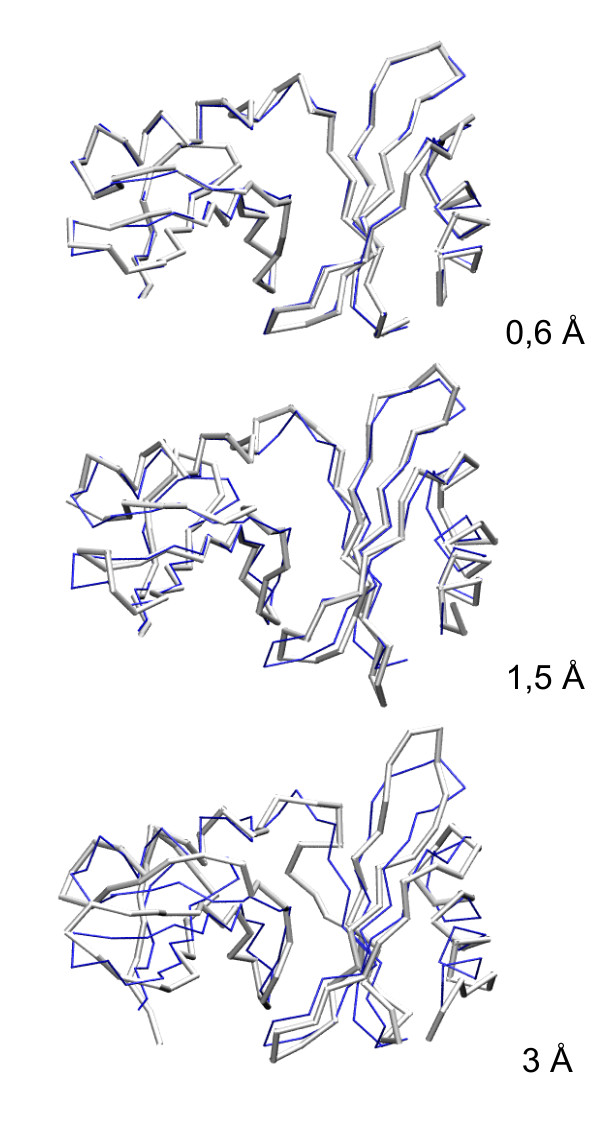
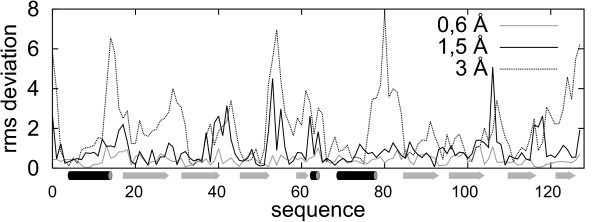
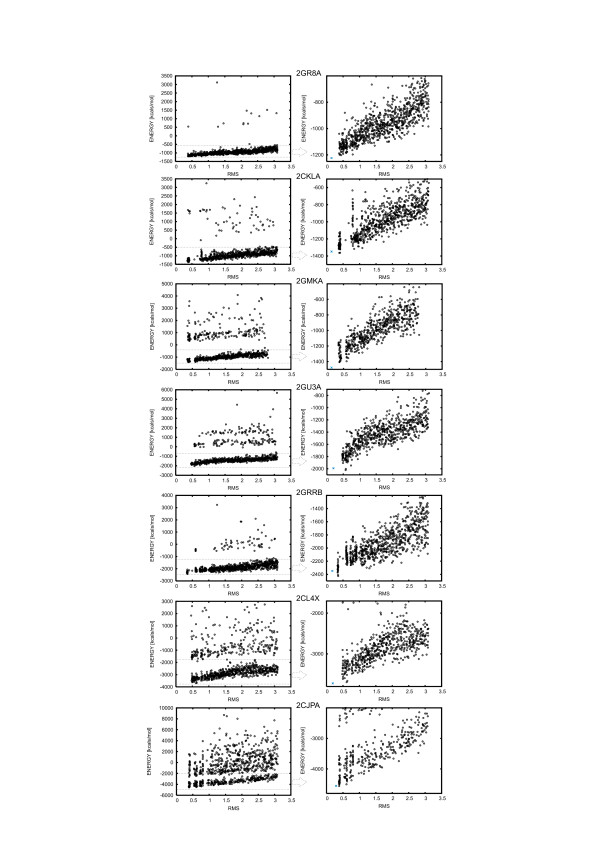
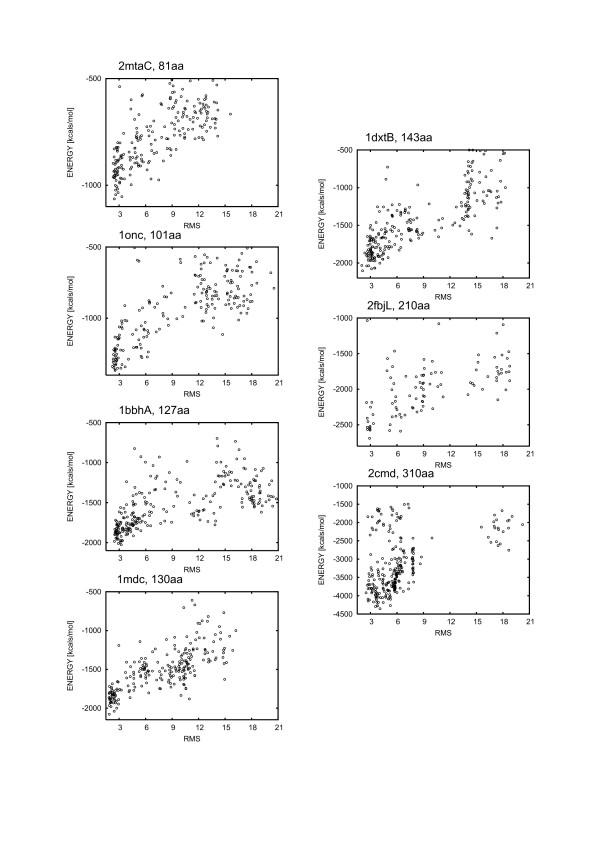
Results: Large sets of structural decoys have been generated using reduced conformational space protein modeling tool CABS. Subsequently, the reduced models were subject to all-atom reconstruction. Then, the resulting detailed models were energy-minimized using state-of-the-art all-atom force field, assuming fixed positions of the alpha carbons. It has been shown that a very short minimization leads to the proper ranking of the quality of the models (distance from the native structure), when the all-atom energy is used as the ranking criterion. Additionally, we performed test on medium and low accuracy decoys built via classical methods of comparative modeling. The test placed our model evaluation procedure among the state-of-the-art protein model assessment methods.
Conclusion: These test computations show that a large scale high resolution protein structure prediction is possible, not only for small but also for large protein domains, and that it should be based on a hierarchical approach to the modeling protocol. We employed Molecular Mechanics with fixed alpha carbons to rank-order the all-atom models built on the scaffolds of the reduced models. Our tests show that a physic-based approach, usually considered computationally too demanding for large-scale applications, can be effectively used in such studies.
Figures
References
-
- Simmerling C, Lee MR, Ortiz AR, Kolinski A, Skolnick J, Kollman PA. Combining MONSSTER and LES/PME to Predict Protein Structure from Amino Acid Sequence: Application to the Small Protein CMTI-1. J Am Chem Soc. 2000;122:8392 –88402. doi: 10.1021/ja993119k. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
