

Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury
- PMID: 17607357
- PMCID: PMC1904311
- DOI: 10.1172/JCI30848
Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury
Abstract
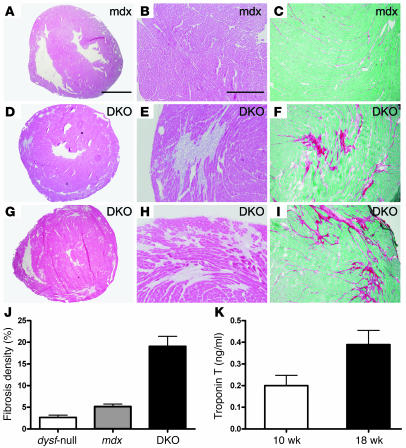
Dilated cardiomyopathy is a life-threatening syndrome that can arise from a myriad of causes, but predisposition toward this malady is inherited in many cases. A number of inherited forms of dilated cardiomyopathy arise from mutations in genes that encode proteins involved in linking the cytoskeleton to the extracellular matrix, and disruption of this link renders the cell membrane more susceptible to injury. Membrane repair is an important cellular mechanism that animal cells have developed to survive membrane disruption. We have previously shown that dysferlin deficiency leads to defective membrane resealing in skeletal muscle and muscle necrosis; however, the function of dysferlin in the heart remains to be determined. Here, we demonstrate that dysferlin is also involved in cardiomyocyte membrane repair and that dysferlin deficiency leads to cardiomyopathy. In particular, stress exercise disturbs left ventricular function in dysferlin-null mice and increases Evans blue dye uptake in dysferlin-deficient cardiomyocytes. Furthermore, a combined deficiency of dystrophin and dysferlin leads to early onset cardiomyopathy. Our results suggest that dysferlin-mediated membrane repair is important for maintaining membrane integrity of cardiomyocytes, particularly under conditions of mechanical stress. Thus, our study establishes what we believe is a novel mechanism underlying the cardiomyopathy that results from a defective membrane repair in the absence of dysferlin.
Figures





Comment in
-
Torn apart: membrane rupture in muscular dystrophies and associated cardiomyopathies.J Clin Invest. 2007 Jul;117(7):1749-52. doi: 10.1172/JCI32686. J Clin Invest. 2007. PMID: 17607350 Free PMC article.
References
-
- Graham R.M., Owens W.A. Pathogenesis of inherited forms of dilated cardiomyopathy. N. Engl. J. Med. 1999;341:1759–1762. - PubMed
-
- Towbin J.A., Bowles N.E. Dilated cardiomyopathy: a tale of cytoskeletal proteins and beyond. J. Cardiovasc. Electrophysiol. 2006;17:919–926. - PubMed
-
- Muntoni F., et al. Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N. Engl. J. Med. 1993;329:921–925. - PubMed
-
- Franz W.M., et al. Association of nonsense mutation of dystrophin gene with disruption of sarcoglycan complex in X-linked dilated cardiomyopathy. Lancet. 2000;355:1781–1785. - PubMed
-
- Piccolo F., et al. Primary adhalinopathy: a common cause of autosomal recessive muscular dystrophy of variable severity. Nat. Genet. 1995;10:243–245. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
