

Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia
- PMID: 17607358
- PMCID: PMC1904315
- DOI: 10.1172/JCI31080
Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia
Abstract
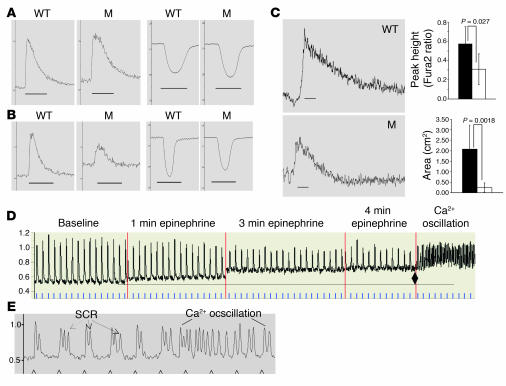
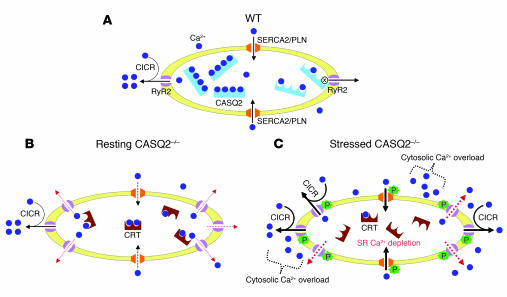
Catecholamine-induced polymorphic ventricular tachycardia (CPVT) is a familial disorder caused by cardiac ryanodine receptor type 2 (RyR2) or calsequestrin 2 (CASQ2) gene mutations. To define how CASQ2 mutations cause CPVT, we produced and studied mice carrying a human D307H missense mutation (CASQ(307/307)) or a CASQ2-null mutation (CASQ(DeltaE9/DeltaE9)). Both CASQ2 mutations caused identical consequences. Young mutant mice had structurally normal hearts but stress-induced ventricular arrhythmias; aging produced cardiac hypertrophy and reduced contractile function. Mutant myocytes had reduced CASQ2 and increased calreticulin and RyR2 (with normal phosphorylated proportions) but unchanged calstabin levels, as well as reduced total sarcoplasmic reticulum (SR) Ca(2+), prolonged Ca(2+) release, and delayed Ca(2+) reuptake. Stress further diminished Ca(2+) transients, elevated cytosolic Ca(2+), and triggered frequent, spontaneous SR Ca(2+) release. Treatment with Mg(2+), a RyR2 inhibitor, normalized myocyte Ca(2+) cycling and decreased CPVT in mutant mice, indicating RyR2 dysfunction was critical to mutant CASQ2 pathophysiology. We conclude that CPVT-causing CASQ2 missense mutations function as null alleles. In the absence of CASQ2, calreticulin, a fetal Ca(2+)-binding protein normally downregulated at birth, remains a prominent SR component. Adaptive changes to CASQ2 deficiency (increased posttranscriptional expression of calreticulin and RyR2) maintained electrical-mechanical coupling, but increased RyR2 leakiness, a paradoxical response further exacerbated by stress. The central role of RyR2 dysfunction in CASQ2 deficiency unifies the pathophysiologic mechanism underlying CPVT due to RyR2 or CASQ2 mutations and suggests a therapeutic approach for these inherited cardiac arrhythmias.
Figures
Comment in
-
Chain-reaction Ca(2+) signaling in the heart.J Clin Invest. 2007 Jul;117(7):1758-62. doi: 10.1172/JCI32496. J Clin Invest. 2007. PMID: 17607353 Free PMC article.
Similar articles
-
Prevention of ventricular arrhythmia and calcium dysregulation in a catecholaminergic polymorphic ventricular tachycardia mouse model carrying calsequestrin-2 mutation.J Cardiovasc Electrophysiol. 2011 Mar;22(3):316-24. doi: 10.1111/j.1540-8167.2010.01877.x. Epub 2010 Aug 30. J Cardiovasc Electrophysiol. 2011. PMID: 20807279 Free PMC article.
-
Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death.Circ Res. 2006 May 12;98(9):1151-8. doi: 10.1161/01.RES.0000220647.93982.08. Epub 2006 Apr 6. Circ Res. 2006. PMID: 16601229
-
The cardiac ryanodine receptor luminal Ca2+ sensor governs Ca2+ waves, ventricular tachyarrhythmias and cardiac hypertrophy in calsequestrin-null mice.Biochem J. 2014 Jul 1;461(1):99-106. doi: 10.1042/BJ20140126. Biochem J. 2014. PMID: 24758151 Free PMC article.
-
Catecholaminergic polymorphic ventricular tachycardia: recent mechanistic insights.Cardiovasc Res. 2005 Aug 15;67(3):379-87. doi: 10.1016/j.cardiores.2005.04.027. Cardiovasc Res. 2005. PMID: 15913575 Review.
-
Molecular basis of catecholaminergic polymorphic ventricular tachycardia.Heart Rhythm. 2009 Jan;6(1):123-9. doi: 10.1016/j.hrthm.2008.09.013. Epub 2008 Sep 16. Heart Rhythm. 2009. PMID: 19121813 Review.
Cited by
-
Integrative Analyses Identify Potential Key Genes and Calcium-Signaling Pathway in Familial Atrioventricular Nodal Reentrant Tachycardia Using Whole-Exome Sequencing.Front Cardiovasc Med. 2022 Jul 18;9:910826. doi: 10.3389/fcvm.2022.910826. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35924220 Free PMC article.
-
It is not always digitalis: bidirectional ventricular tachycardia in left ventricular hypertrophy.Ann Noninvasive Electrocardiol. 2008 Apr;13(2):208-10. doi: 10.1111/j.1542-474X.2008.00218.x. Ann Noninvasive Electrocardiol. 2008. PMID: 18426446 Free PMC article. No abstract available.
-
Quantitative proteomics for cardiac biomarker discovery using isoproterenol-treated nonhuman primates.J Proteome Res. 2014 Dec 5;13(12):5909-17. doi: 10.1021/pr500835w. Epub 2014 Nov 7. J Proteome Res. 2014. PMID: 25345801 Free PMC article.
-
Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease.Biomolecules. 2022 Jul 26;12(8):1030. doi: 10.3390/biom12081030. Biomolecules. 2022. PMID: 35892340 Free PMC article. Review.
-
Alpha blockade potentiates CPVT therapy in calsequestrin-mutant mice.Heart Rhythm. 2014 Aug;11(8):1471-9. doi: 10.1016/j.hrthm.2014.04.030. Epub 2014 Apr 21. Heart Rhythm. 2014. PMID: 24768611 Free PMC article.
References
-
- Leenhardt A., et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519. - PubMed
-
- Priori S.G., et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. - PubMed
-
- Lahat H., et al. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13-21. Circulation. 2001;103:2822–2827. - PubMed
-
- Postma A.V., et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2002;91:e21–e26. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
