

QM/MM linear response method distinguishes ligand affinities for closely related metalloproteins
- PMID: 17607744
- PMCID: PMC2896063
- DOI: 10.1002/prot.21500
QM/MM linear response method distinguishes ligand affinities for closely related metalloproteins
Abstract
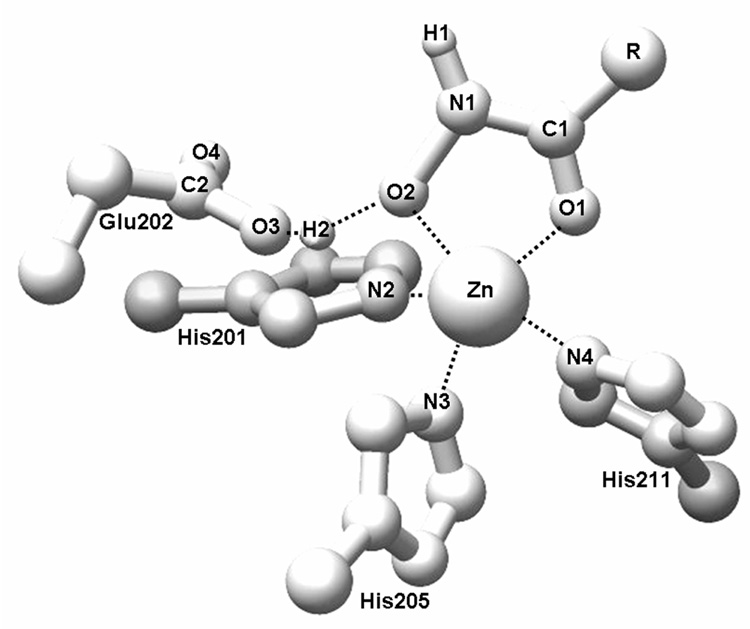
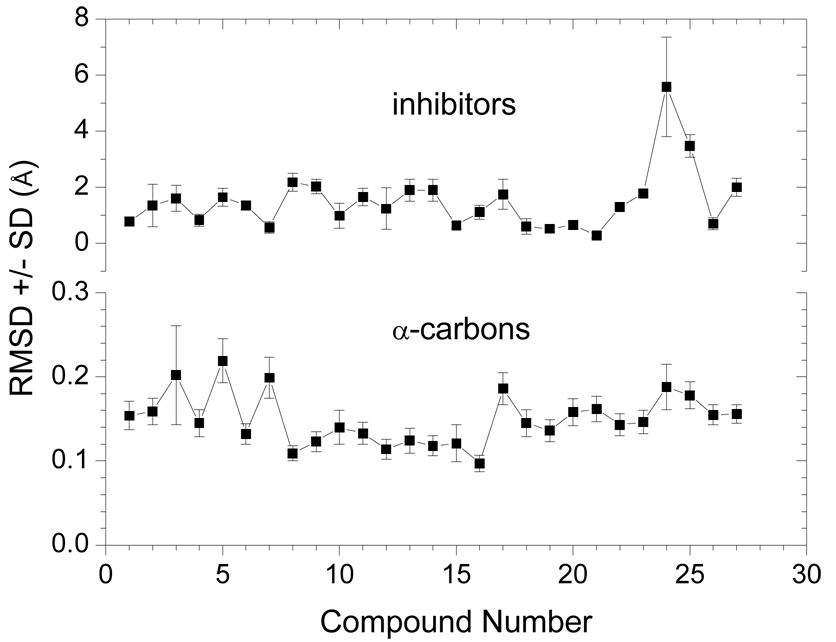
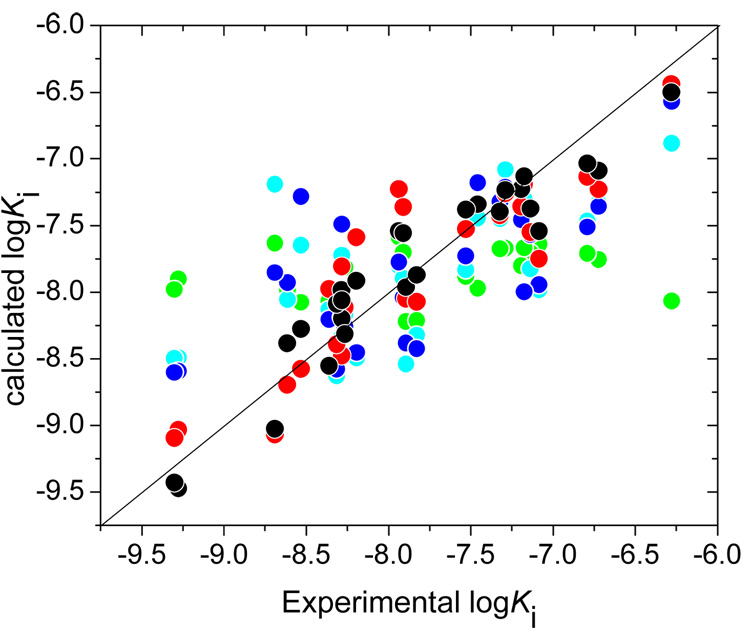
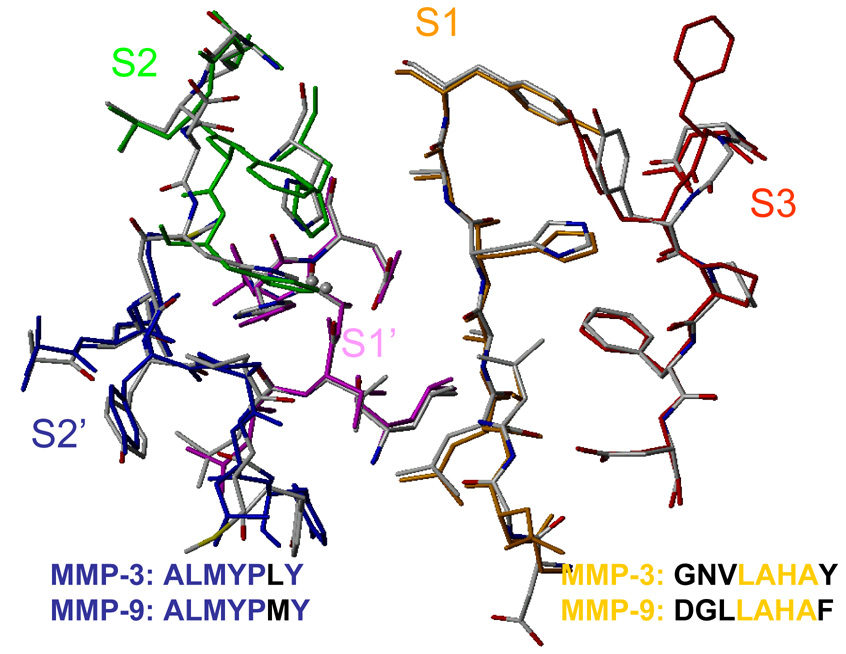
Design of selective ligands for closely related targets is becoming one of the most important tasks in the drug development. New tools, more precise than fast scoring functions and less demanding than sophisticated Free Energy Perturbation methods, are necessary to help accomplish this goal. The methods of intermediate complexity, characterizing individual contributions to the binding energy, have been an area of intense research in the past few years. Our recently developed quantum mechanical/molecular mechanical (QM/MM) modification of the Linear Response (LR) method describes the binding free energies as the sum of empirically weighted contributions of the QM/MM interaction energies and solvent-accessible surface areas for the time-averaged structures of hydrated complexes, obtained by molecular dynamics (MD) simulations. The method was applied to published data on 27 inhibitors of matrix metalloproteinase-3 (MMP-3). The two descriptors explained 90% of variance in the inhibition constants with RMSE of 0.245 log units. The QM/MM treatment is indispensable for characterization of the systems lacking suitable force-field expressions. In this case, it provided characteristics of H-bonds of the inhibitors to Glu202, charges of binding site atoms, and accurate coordination geometries of the ligands to catalytic zinc. The geometries were constrained during the MD simulations, which characterized conformational flexibility of the complexes and helped in the elucidation of the binding differences for related compounds. A comparison of the presented QM/MM LR results with those previously published for inhibition of MMP-9 by the same set of ligands showed that the QM/MM LR approach was able to distinguish subtle differences in binding affinities for MMP-3 and MMP-9, which did not exceed one order of magnitude. This precision level makes the approach a useful tool for design of selective ligands to similar targets, because the results can be safely extrapolated to maximize selectivity.
(c) 2007 Wiley-Liss, Inc.
Figures
Similar articles
-
A combination of docking, QM/MM methods, and MD simulation for binding affinity estimation of metalloprotein ligands.J Med Chem. 2005 Aug 25;48(17):5437-47. doi: 10.1021/jm049050v. J Med Chem. 2005. PMID: 16107143 Free PMC article.
-
Improved estimation of ligand-macromolecule binding affinities by linear response approach using a combination of multi-mode MD simulation and QM/MM methods.J Comput Aided Mol Des. 2007 Jan-Mar;21(1-3):131-7. doi: 10.1007/s10822-007-9104-4. Epub 2007 Feb 28. J Comput Aided Mol Des. 2007. PMID: 17333483 Free PMC article.
-
Computation of Protein-Ligand Binding Free Energies with a Quantum Mechanics-Based Mining Minima Algorithm.J Chem Theory Comput. 2025 Apr 22;21(8):4236-4265. doi: 10.1021/acs.jctc.4c01707. Epub 2025 Apr 9. J Chem Theory Comput. 2025. PMID: 40202178 Free PMC article.
-
Advances in binding free energies calculations: QM/MM-based free energy perturbation method for drug design.Curr Pharm Des. 2013;19(26):4674-86. doi: 10.2174/1381612811319260002. Curr Pharm Des. 2013. PMID: 23260025 Review.
-
Calculations on noncovalent interactions and databases of benchmark interaction energies.Acc Chem Res. 2012 Apr 17;45(4):663-72. doi: 10.1021/ar200255p. Epub 2012 Jan 6. Acc Chem Res. 2012. PMID: 22225511 Review.
Cited by
-
QM/MM Studies of the Matrix Metalloproteinase 2 (MMP2) Inhibition Mechanism of (S)-SB-3CT and its Oxirane Analogue.J Chem Theory Comput. 2010 Nov 9;6(11):3580-3587. doi: 10.1021/ct100382k. J Chem Theory Comput. 2010. PMID: 21076643 Free PMC article.
-
Matrix metalloproteinase 2 inhibition: combined quantum mechanics and molecular mechanics studies of the inhibition mechanism of (4-phenoxyphenylsulfonyl)methylthiirane and its oxirane analogue.Biochemistry. 2009 Oct 20;48(41):9839-47. doi: 10.1021/bi901118r. Biochemistry. 2009. PMID: 19754151 Free PMC article.
-
Rigorous incorporation of tautomers, ionization species, and different binding modes into ligand-based and receptor-based 3D-QSAR methods.Curr Pharm Des. 2013;19(23):4316-22. doi: 10.2174/1381612811319230013. Curr Pharm Des. 2013. PMID: 23170882 Free PMC article. Review.
-
Mechanisms of Proteolytic Enzymes and Their Inhibition in QM/MM Studies.Int J Mol Sci. 2021 Mar 22;22(6):3232. doi: 10.3390/ijms22063232. Int J Mol Sci. 2021. PMID: 33810118 Free PMC article. Review.
-
Binding affinity prediction for ligands and receptors forming tautomers and ionization species: inhibition of mitogen-activated protein kinase-activated protein kinase 2 (MK2).J Med Chem. 2012 Mar 8;55(5):2035-47. doi: 10.1021/jm201217q. Epub 2012 Feb 17. J Med Chem. 2012. PMID: 22280316 Free PMC article.
References
-
- Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: Current status and future challenges. Proteins. 2006;65:15–26. - PubMed
-
- Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. - PubMed
-
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. - PubMed
-
- Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. - PubMed
-
- Bohm HJ. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J Comput Aided Mol Des. 1994;8:243–256. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
