

Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway
- PMID: 17609269
- PMCID: PMC2045390
- DOI: 10.1128/JVI.00541-07
Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway
Abstract
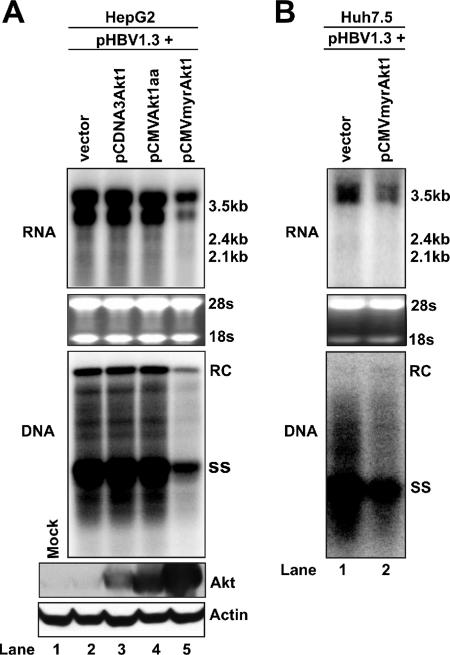
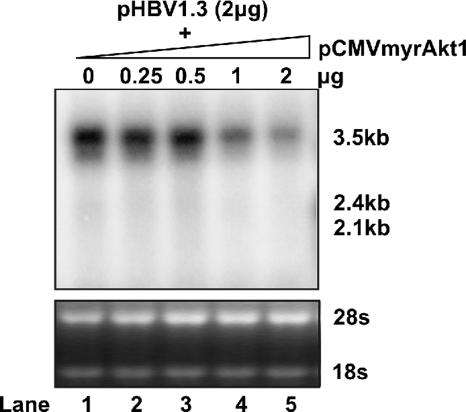
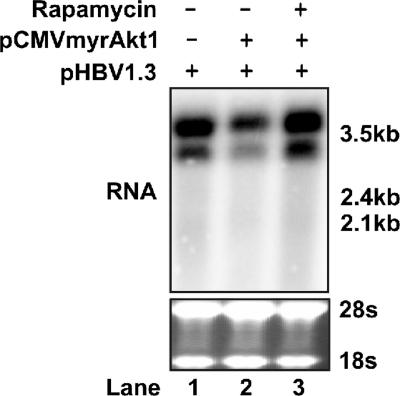
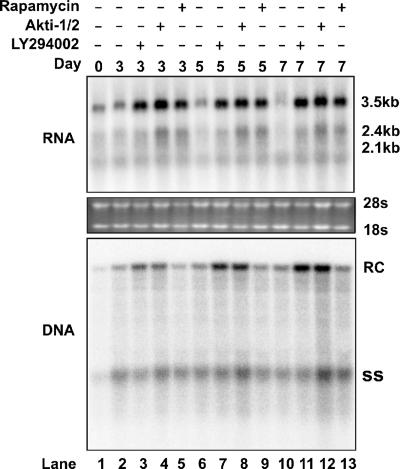
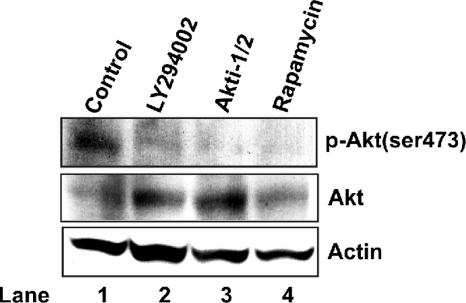
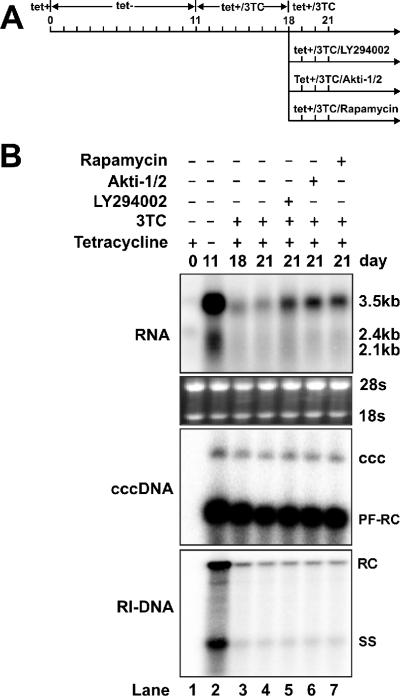
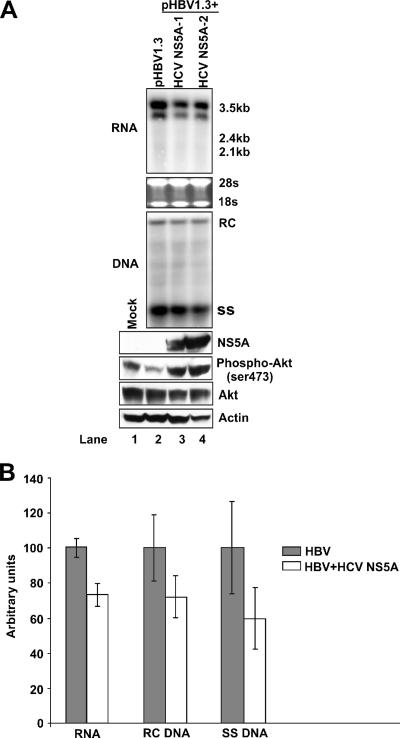
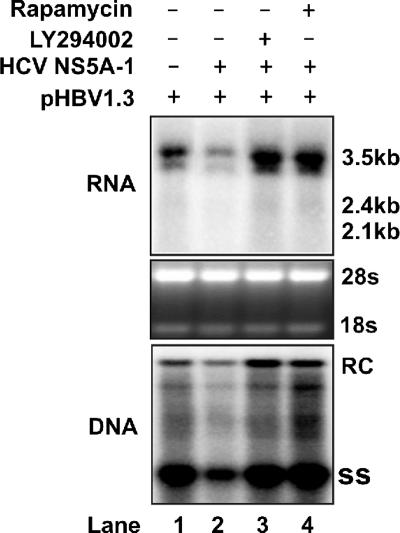
The phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt) signaling pathway is one of the major oncogenic pathways and is activated in many types of human cancers, including hepatocellular carcinoma. It can also be activated by the hepatitis C virus (HCV) nonstructural 5A (NS5A) protein. In the present study, we set out to determine the regulatory effects of this pathway on the replication of hepatitis B virus (HBV). Our results demonstrate that the expression of a constitutively active Akt1 profoundly inhibited HBV RNA transcription and consequently reduced HBV DNA replication in HepG2 cells. This suppression of HBV gene transcription was apparently mediated by the activation of mTOR, as it was abolished by the mTOR inhibitor rapamycin. Moreover, treatment of HBV-expressing HepG2.2.15 cells with inhibitors of PI3K, Akt, and mTOR increased the transcription of 3.5-kb and 2.4-kb viral RNA as well as the replication of HBV DNA. This observation implies that the basal level activation of this pathway in HepG2 cells regulated HBV replication. Consistent with previous reports showing that the HCV NS5A protein could bind to the p85 subunit of PI3K and activate the PI3K-Akt signal transduction pathway, our results showed that expression of this protein could inhibit HBV RNA transcription and reduce HBV DNA replication in HepG2 cells. Taken together, our results suggest that the activation of the PI3K-Akt pathway during liver oncogenesis may be at least partially responsible for the elimination of HBV replication from tumor cells and may also provide an explanation for the observed suppression of HBV replication by HCV coinfection.
Figures
References
-
- Barnett, S. F., D. Defeo-Jones, S. Fu, P. J. Hancock, K. M. Haskell, R. E. Jones, J. A. Kahana, A. M. Kral, K. Leander, L. L. Lee, J. Malinowski, E. M. McAvoy, D. D. Nahas, R. G. Robinson, and H. E. Huber. 2005. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 385:399-408. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
