

Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons
- PMID: 17611284
- PMCID: PMC6794597
- DOI: 10.1523/JNEUROSCI.1831-07.2007
Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons
Abstract
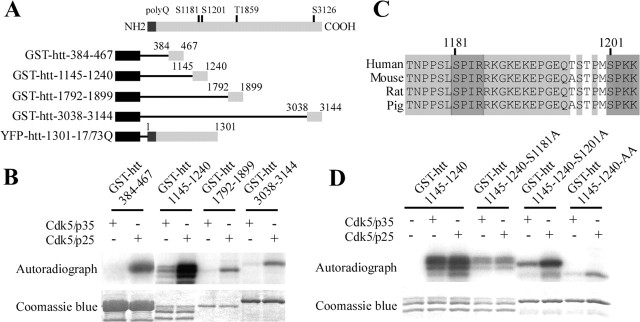
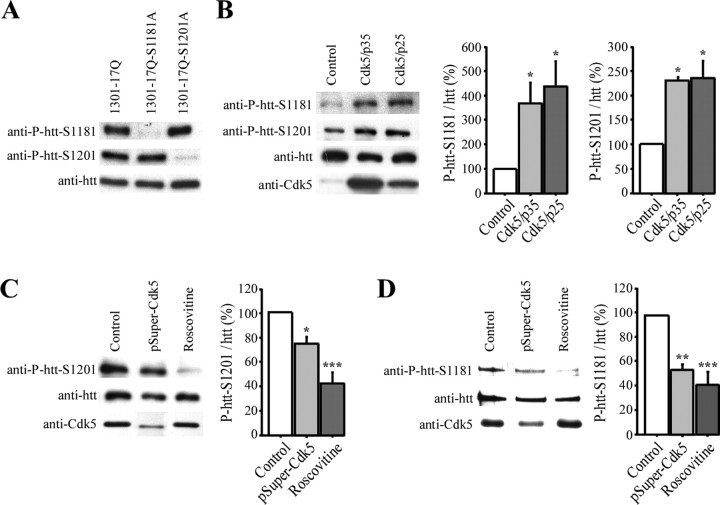
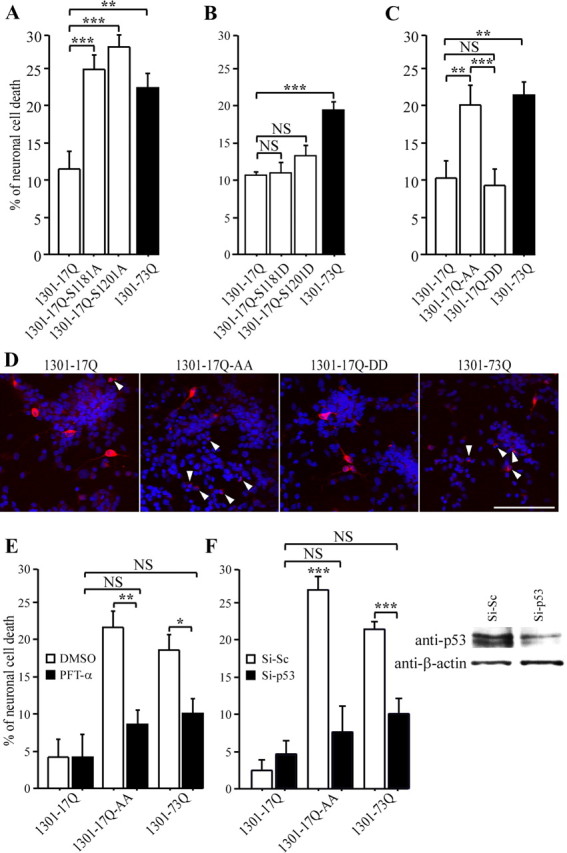
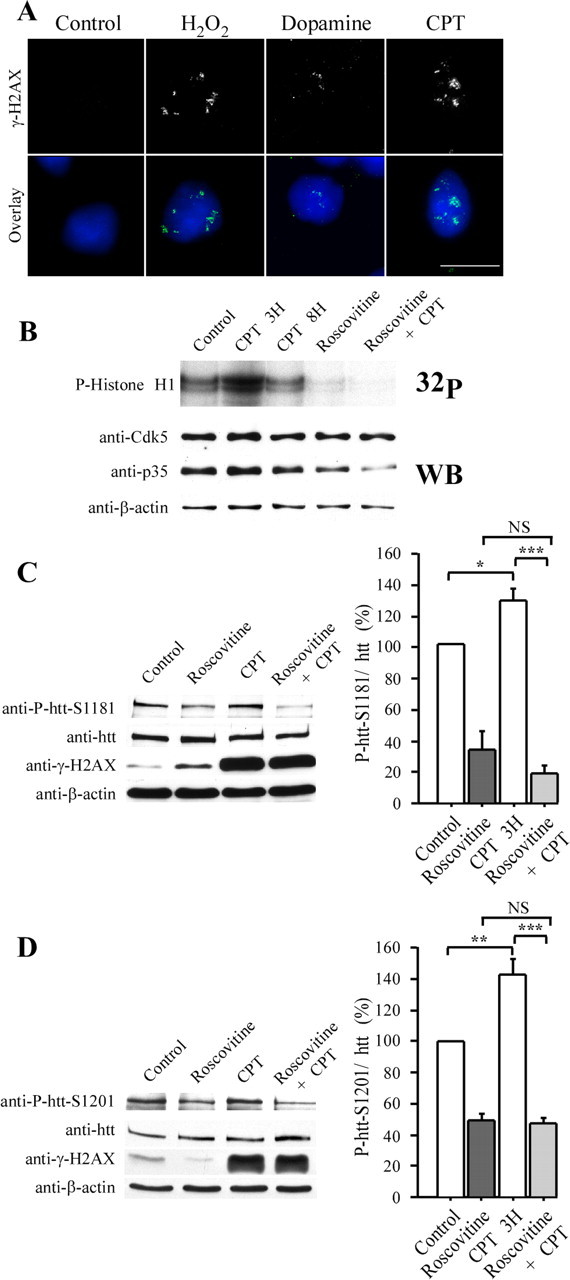
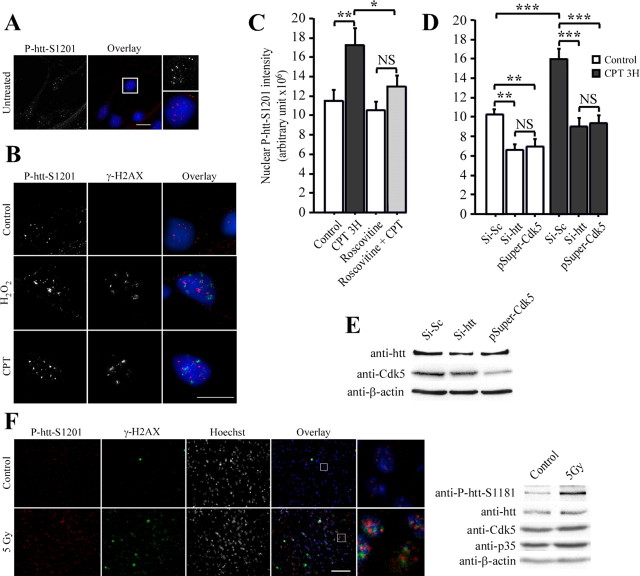
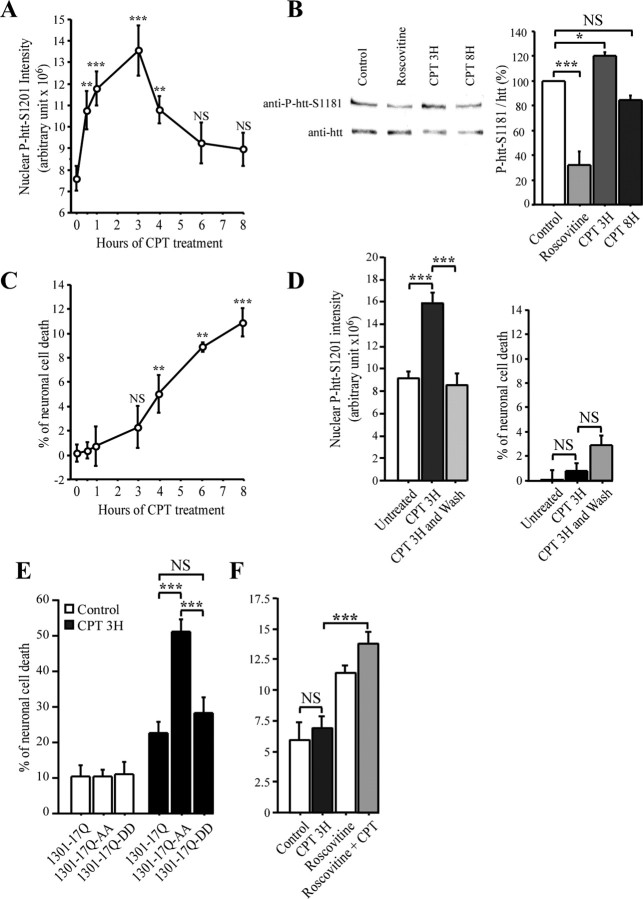
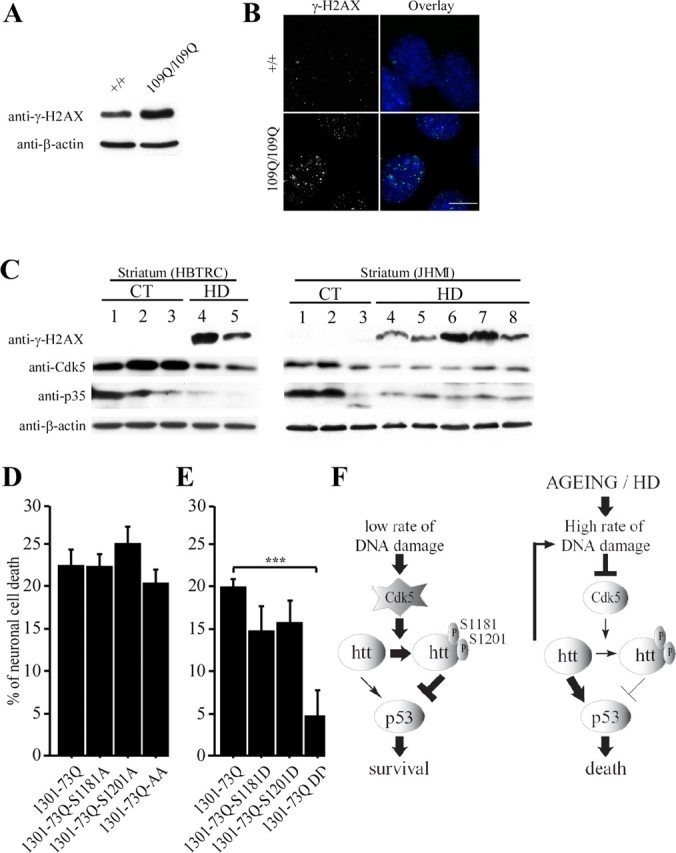
Huntingtin is an antiapoptotic protein that becomes toxic when its polyglutamine stretch is expanded, resulting in Huntington's disease (HD). Protein context and posttranslational modifications regulate huntingtin toxicity. Identifying signaling pathways that act on huntingtin is, therefore, key to understanding huntingtin function in normal and pathological conditions. We show here that huntingtin is phosphorylated by the cyclin-dependent kinase 5 (Cdk5) at serines 1181 and 1201. Phosphorylation can be induced by DNA damage in vitro and in vivo. The state of huntingtin phosphorylation is a crucial regulator of neuronal cell death. Absence of phosphorylation of huntingtin at serines 1181 and 1201 confers toxic properties to wild-type huntingtin in a p53-dependent manner in striatal neurons and accelerates neuronal death induced by DNA damage. In contrast, phosphorylation at serines 1181 and 1201 protects against polyQ-induced toxicity. Finally, we show in late stages of HD a sustained DNA damage that is associated with a decrease in Cdk5/p35 levels. We propose that wild-type huntingtin is a component of the DNA damage response signal in neurons and that the Cdk5/DNA damage pathway is dysregulated in HD.
Figures
References
-
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. - PubMed
-
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. - PMC - PubMed
-
- Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. - PubMed
-
- Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington's disease. J Neurochem. 2001;79:1246–1249. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous