

Leveraging hierarchical population structure in discrete association studies
- PMID: 17611623
- PMCID: PMC1899226
- DOI: 10.1371/journal.pone.0000591
Leveraging hierarchical population structure in discrete association studies
Abstract
Population structure can confound the identification of correlations in biological data. Such confounding has been recognized in multiple biological disciplines, resulting in a disparate collection of proposed solutions. We examine several methods that correct for confounding on discrete data with hierarchical population structure and identify two distinct confounding processes, which we call coevolution and conditional influence. We describe these processes in terms of generative models and show that these generative models can be used to correct for the confounding effects. Finally, we apply the models to three applications: identification of escape mutations in HIV-1 in response to specific HLA-mediated immune pressure, prediction of coevolving residues in an HIV-1 peptide, and a search for genotypes that are associated with bacterial resistance traits in Arabidopsis thaliana. We show that coevolution is a better description of confounding in some applications and conditional influence is better in others. That is, we show that no single method is best for addressing all forms of confounding. Analysis tools based on these models are available on the internet as both web based applications and downloadable source code at http://atom.research.microsoft.com/bio/phylod.aspx.
Conflict of interest statement
Figures
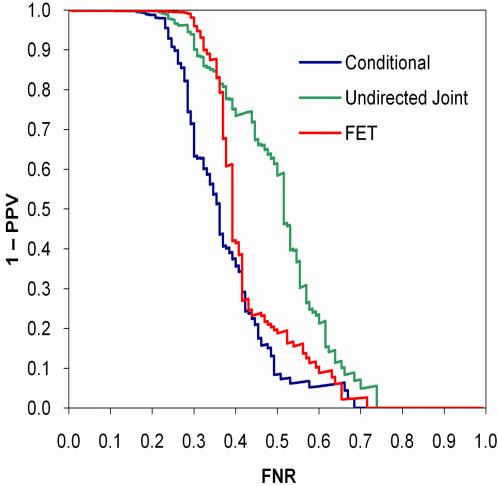
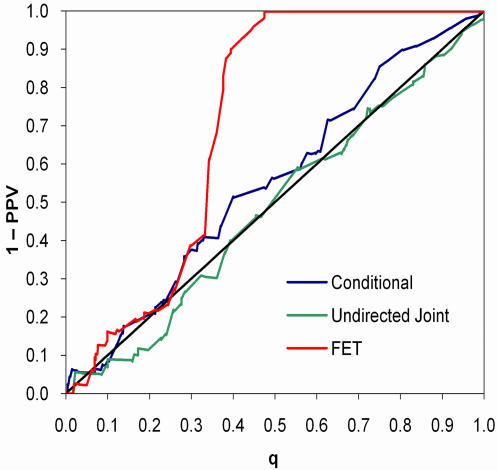
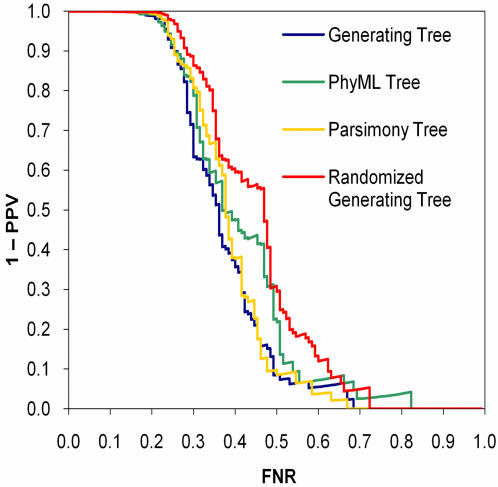
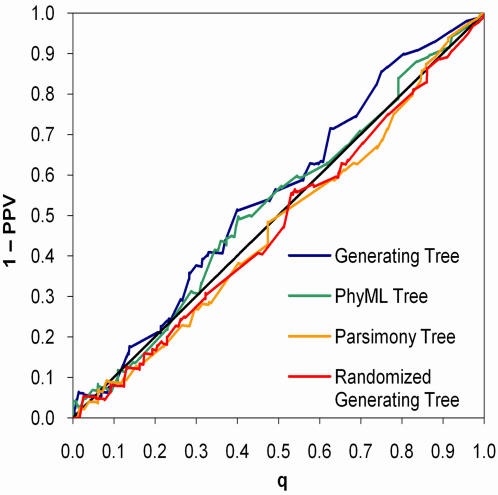
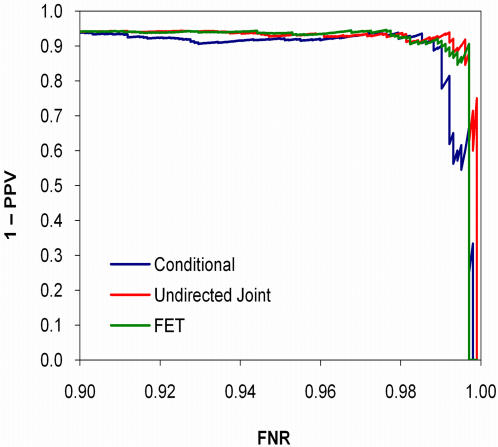
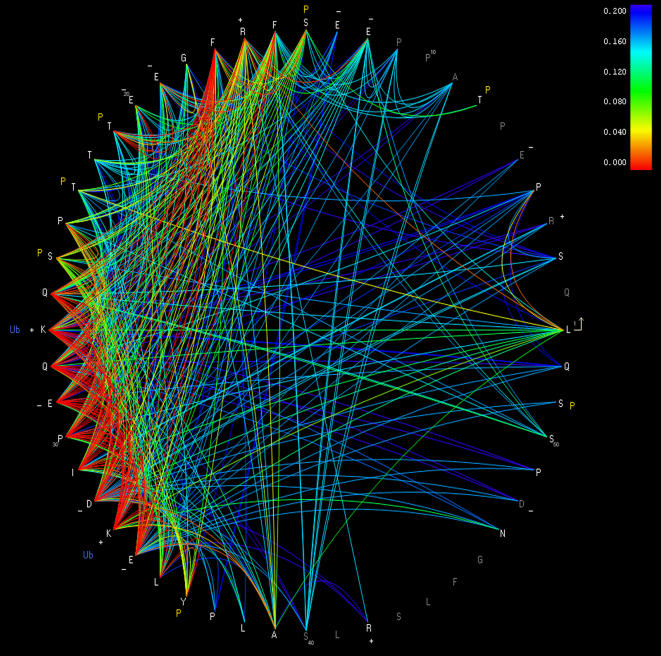
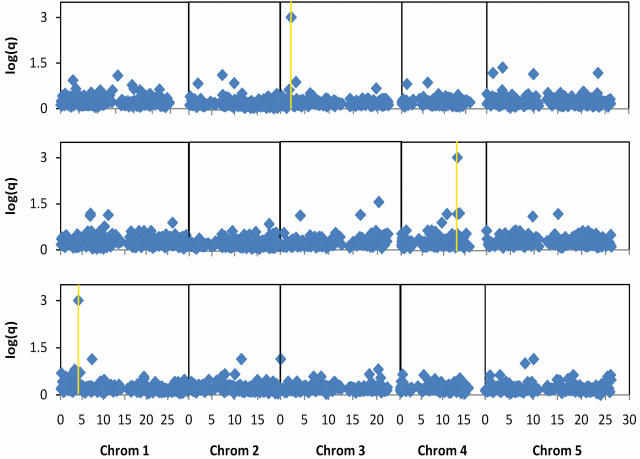
References
-
- Felsenstein Phylogenies and the comparative method. American Naturalist. 1985;125 - PubMed
-
- Ridley M. The Explanation of Organic Diversity: The Comparative Method and Adaptations for Mating. Oxford: Oxford University Press; 1983.
-
- Maddison DR. 1990. Phylogenetic Inference of Historical Pathways and Models of Evolutionary Change. Ph.D. thesis, Harvard University, Cambridge, MA.
-
- Pagel M. Detecting correlated evolution on phylogenies: a general method for the comparative analysis of discrete characters. Proc R Soc Lond B Biol Sci. 1994;255:37–45.
-
- Pollock DD, Taylor WR, Goldman N. Coevolving protein residues: maximum likelihood identification and relationship to structure. J Mol Biol. 1999;287:187–198. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
