

A proteome-wide protein interaction map for Campylobacter jejuni
- PMID: 17615063
- PMCID: PMC2323224
- DOI: 10.1186/gb-2007-8-7-r130
A proteome-wide protein interaction map for Campylobacter jejuni
Abstract
Background: Data from large-scale protein interaction screens for humans and model eukaryotes have been invaluable for developing systems-level models of biological processes. Despite this value, only a limited amount of interaction data is available for prokaryotes. Here we report the systematic identification of protein interactions for the bacterium Campylobacter jejuni, a food-borne pathogen and a major cause of gastroenteritis worldwide.
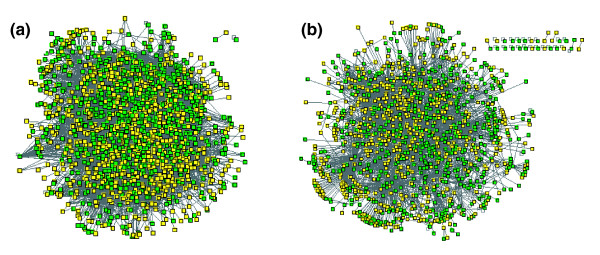
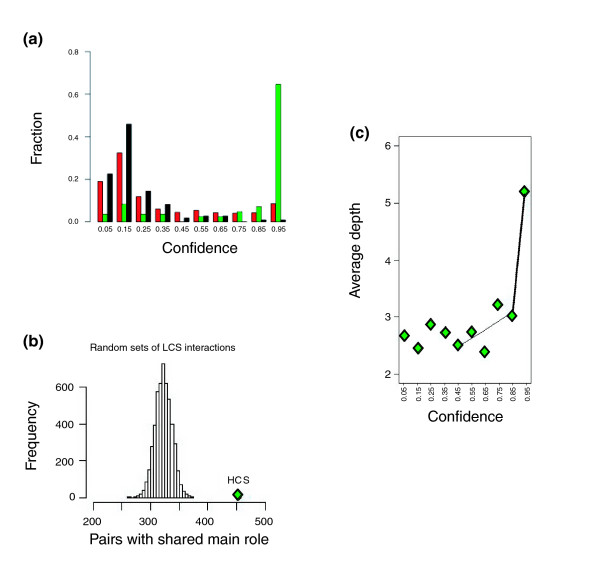
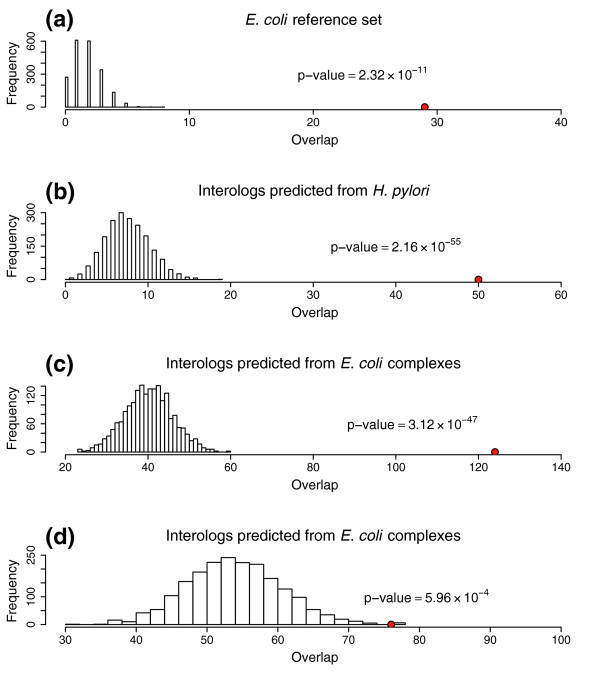
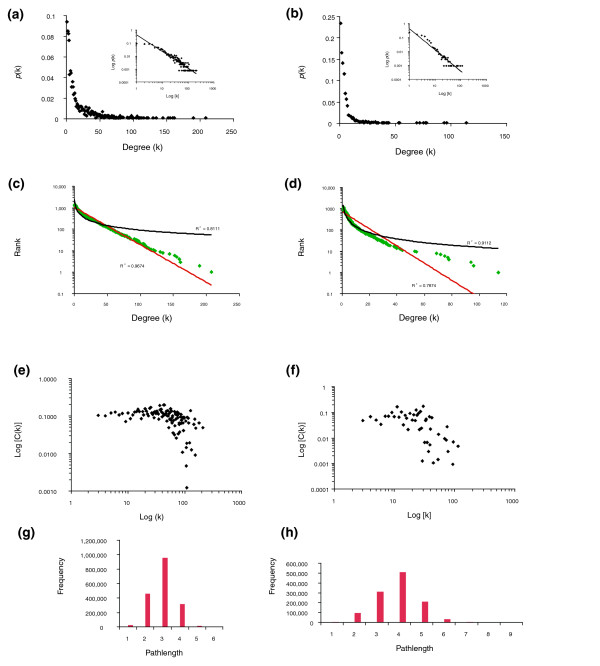
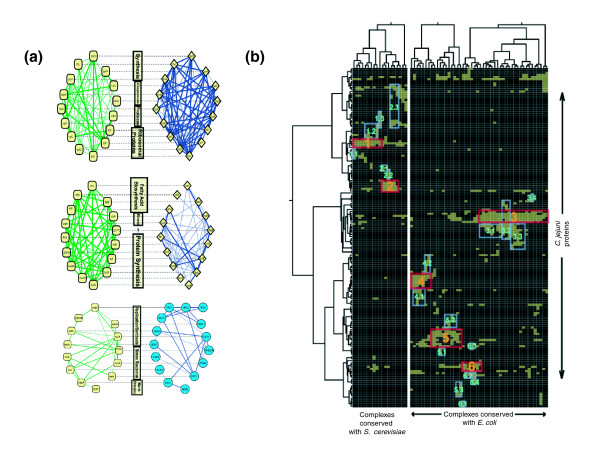
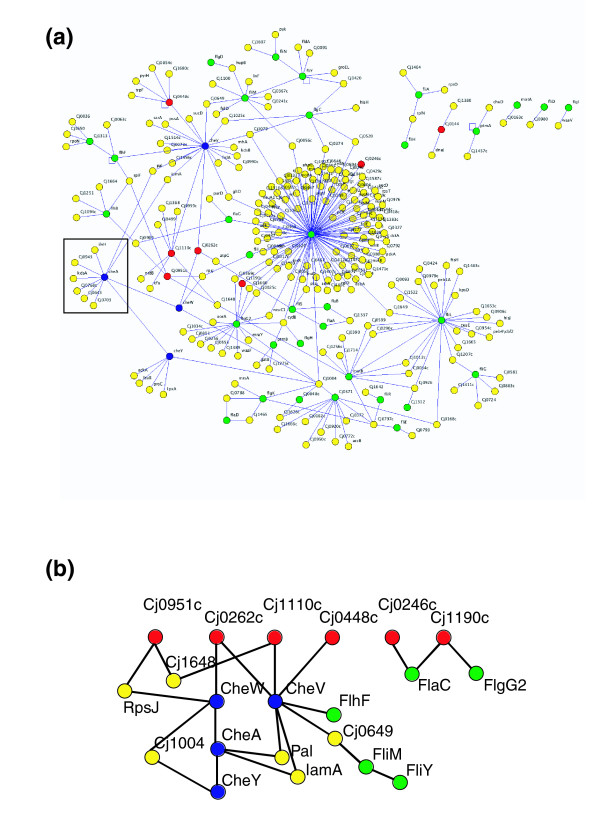
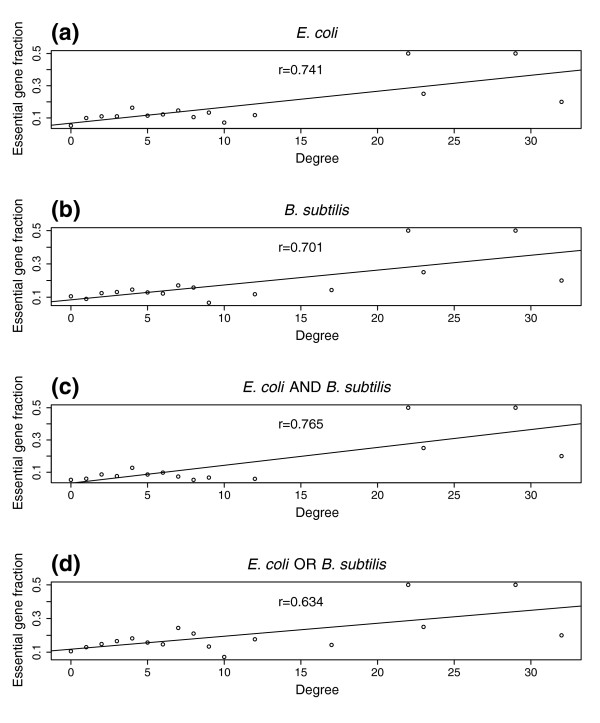
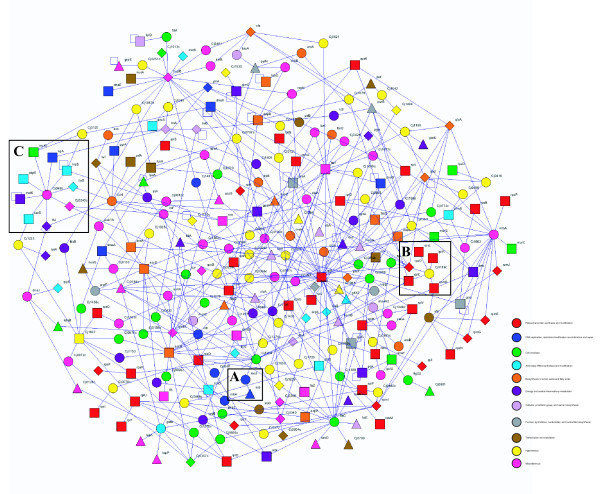
Results: Using high-throughput yeast two-hybrid screens we detected and reproduced 11,687 interactions. The resulting interaction map includes 80% of the predicted C. jejuni NCTC11168 proteins and places a large number of poorly characterized proteins into networks that provide initial clues about their functions. We used the map to identify a number of conserved subnetworks by comparison to protein networks from Escherichia coli and Saccharomyces cerevisiae. We also demonstrate the value of the interactome data for mapping biological pathways by identifying the C. jejuni chemotaxis pathway. Finally, the interaction map also includes a large subnetwork of putative essential genes that may be used to identify potential new antimicrobial drug targets for C. jejuni and related organisms.
Conclusion: The C. jejuni protein interaction map is one of the most comprehensive yet determined for a free-living organism and nearly doubles the binary interactions available for the prokaryotic kingdom. This high level of coverage facilitates pathway mapping and function prediction for a large number of C. jejuni proteins as well as orthologous proteins from other organisms. The broad coverage also facilitates cross-species comparisons for the identification of evolutionarily conserved subnetworks of protein interactions.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
