

Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1
- PMID: 17616974
- PMCID: PMC1904471
- DOI: 10.1371/journal.ppat.0030094
Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1
Erratum in
- PLoS Pathog. 2007 Aug 10;3(8):e121
Abstract
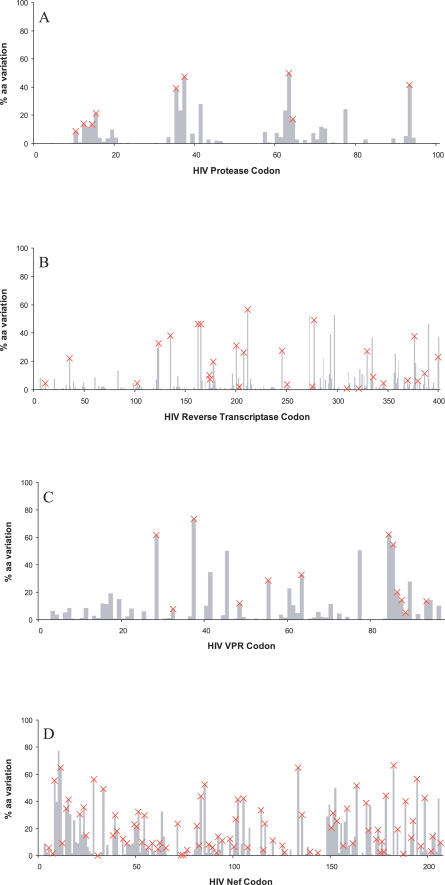
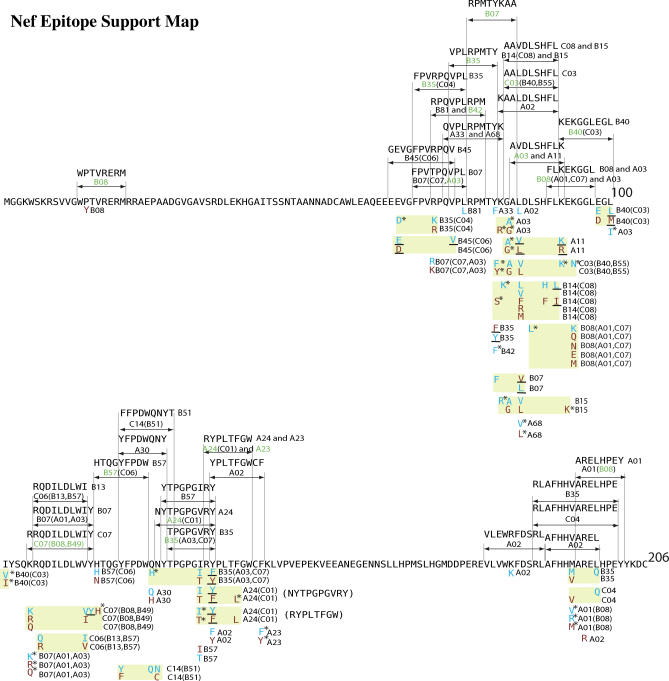
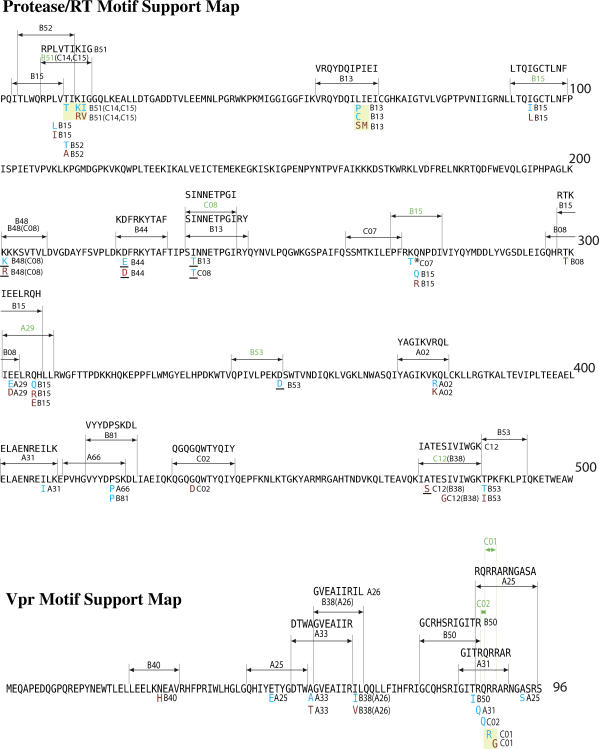
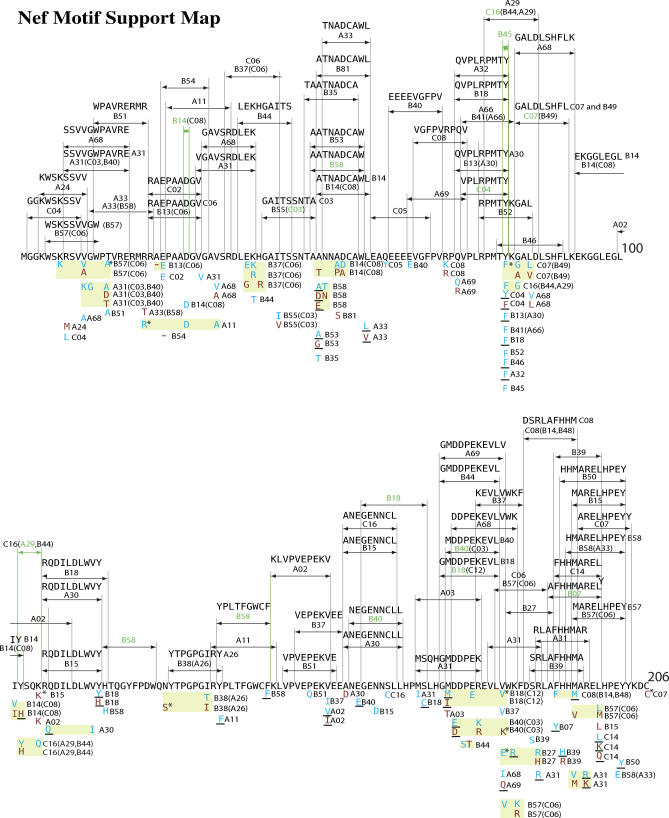
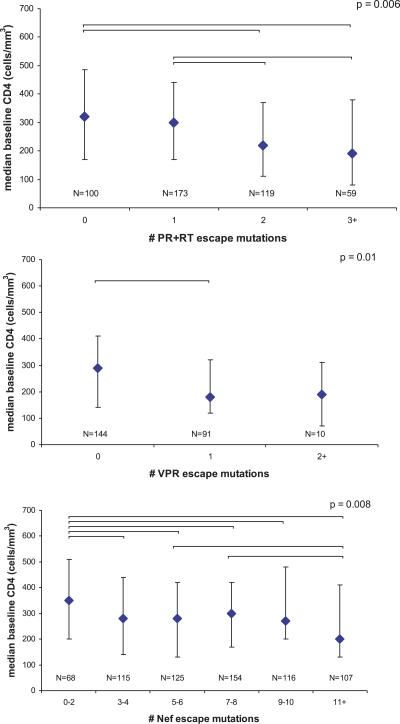
Despite the formidable mutational capacity and sequence diversity of HIV-1, evidence suggests that viral evolution in response to specific selective pressures follows generally predictable mutational pathways. Population-based analyses of clinically derived HIV sequences may be used to identify immune escape mutations in viral genes; however, prior attempts to identify such mutations have been complicated by the inability to discriminate active immune selection from virus founder effects. Furthermore, the association between mutations arising under in vivo immune selection and disease progression for highly variable pathogens such as HIV-1 remains incompletely understood. We applied a viral lineage-corrected analytical method to investigate HLA class I-associated sequence imprinting in HIV protease, reverse transcriptase (RT), Vpr, and Nef in a large cohort of chronically infected, antiretrovirally naïve individuals. A total of 478 unique HLA-associated polymorphisms were observed and organized into a series of "escape maps," which identify known and putative cytotoxic T lymphocyte (CTL) epitopes under selection pressure in vivo. Our data indicate that pathways to immune escape are predictable based on host HLA class I profile, and that epitope anchor residues are not the preferred sites of CTL escape. Results reveal differential contributions of immune imprinting to viral gene diversity, with Nef exhibiting far greater evidence for HLA class I-mediated selection compared to other genes. Moreover, these data reveal a significant, dose-dependent inverse correlation between HLA-associated polymorphisms and HIV disease stage as estimated by CD4(+) T cell count. Identification of specific sites and patterns of HLA-associated polymorphisms across HIV protease, RT, Vpr, and Nef illuminates regions of the genes encoding these products under active immune selection pressure in vivo. The high density of HLA-associated polymorphisms in Nef compared to other genes investigated indicates differential HLA class I-driven evolution in different viral genes. The relationship between HLA class I-associated polymorphisms and lower CD4(+) cell count suggests that immune escape correlates with disease status, supporting an essential role of maintenance of effective CTL responses in immune control of HIV-1. The design of preventative and therapeutic CTL-based vaccine approaches could incorporate information on predictable escape pathways.
Conflict of interest statement
Figures


References
-
- Klein J, Sato A. The HLA system. First of two parts. N Engl J Med. 2000;343:702–709. - PubMed
-
- Moore CB, John M, James IR, Christiansen FT, Witt CS, et al. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 2002;296:1439–1443. - PubMed
-
- Goulder PJ, Brander C, Tang Y, Tremblay C, Colbert RA, et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 2001;412:334–338. - PubMed
-
- Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature. 1991;354:453–459. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
