

Genetic association mapping via evolution-based clustering of haplotypes
- PMID: 17616979
- PMCID: PMC1913101
- DOI: 10.1371/journal.pgen.0030111
Genetic association mapping via evolution-based clustering of haplotypes
Abstract
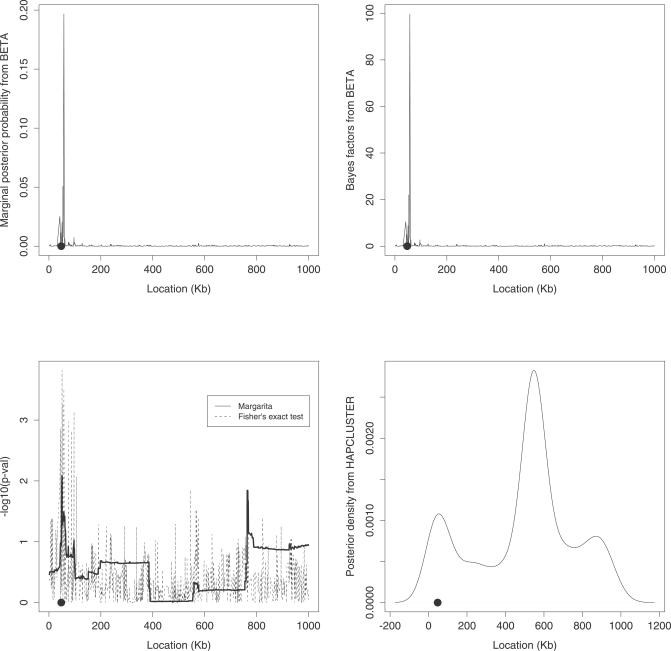
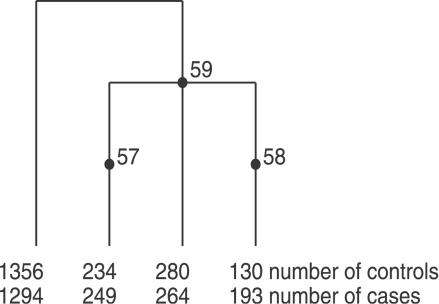
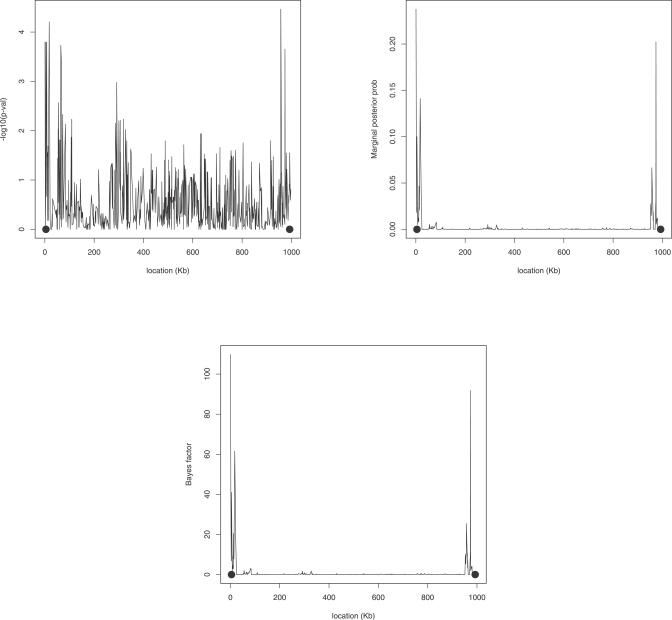
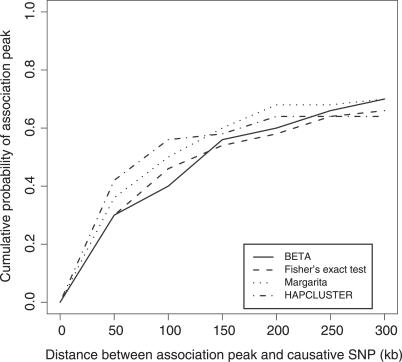
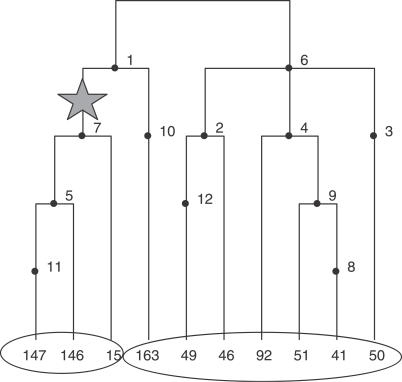
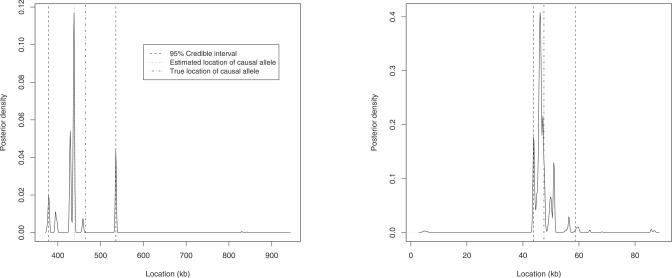
Multilocus analysis of single nucleotide polymorphism haplotypes is a promising approach to dissecting the genetic basis of complex diseases. We propose a coalescent-based model for association mapping that potentially increases the power to detect disease-susceptibility variants in genetic association studies. The approach uses Bayesian partition modelling to cluster haplotypes with similar disease risks by exploiting evolutionary information. We focus on candidate gene regions with densely spaced markers and model chromosomal segments in high linkage disequilibrium therein assuming a perfect phylogeny. To make this assumption more realistic, we split the chromosomal region of interest into sub-regions or windows of high linkage disequilibrium. The haplotype space is then partitioned into disjoint clusters, within which the phenotype-haplotype association is assumed to be the same. For example, in case-control studies, we expect chromosomal segments bearing the causal variant on a common ancestral background to be more frequent among cases than controls, giving rise to two separate haplotype clusters. The novelty of our approach arises from the fact that the distance used for clustering haplotypes has an evolutionary interpretation, as haplotypes are clustered according to the time to their most recent common ancestor. Our approach is fully Bayesian and we develop a Markov Chain Monte Carlo algorithm to sample efficiently over the space of possible partitions. We compare the proposed approach to both single-marker analyses and recently proposed multi-marker methods and show that the Bayesian partition modelling performs similarly in localizing the causal allele while yielding lower false-positive rates. Also, the method is computationally quicker than other multi-marker approaches. We present an application to real genotype data from the CYP2D6 gene region, which has a confirmed role in drug metabolism, where we succeed in mapping the location of the susceptibility variant within a small error.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures
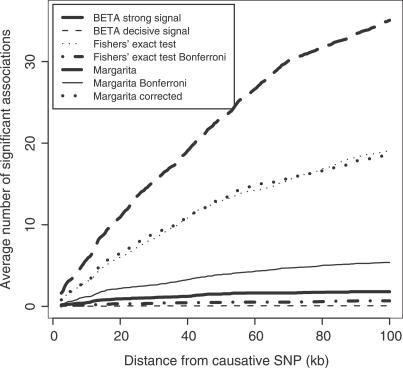
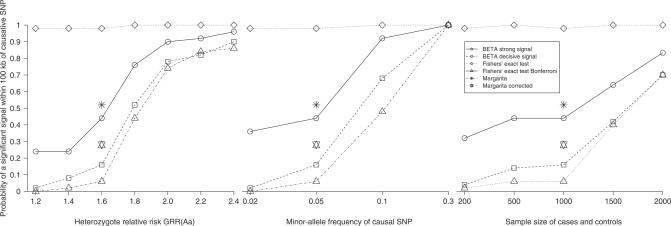
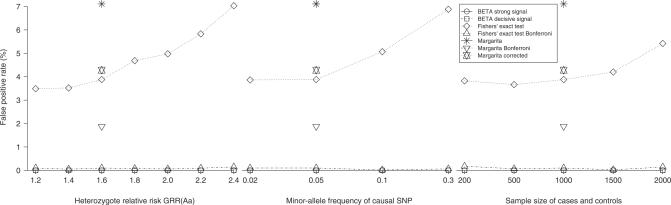
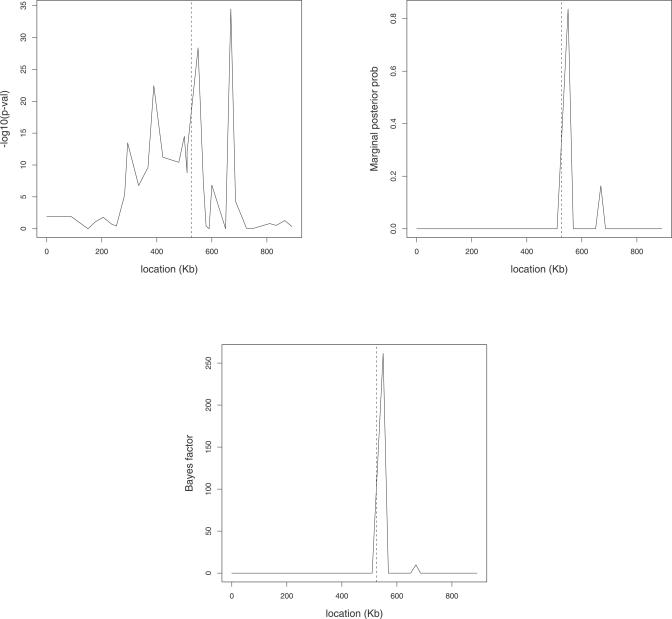
Similar articles
-
Direct analysis of unphased SNP genotype data in population-based association studies via Bayesian partition modelling of haplotypes.Genet Epidemiol. 2005 Sep;29(2):91-107. doi: 10.1002/gepi.20080. Genet Epidemiol. 2005. PMID: 15940704
-
A flexible Bayesian framework for modeling haplotype association with disease, allowing for dominance effects of the underlying causative variants.Am J Hum Genet. 2006 Oct;79(4):679-94. doi: 10.1086/508264. Epub 2006 Aug 31. Am J Hum Genet. 2006. PMID: 16960804 Free PMC article.
-
Fine mapping of disease genes via haplotype clustering.Genet Epidemiol. 2006 Feb;30(2):170-9. doi: 10.1002/gepi.20134. Genet Epidemiol. 2006. PMID: 16385468
-
Tag SNP selection for association studies.Genet Epidemiol. 2004 Dec;27(4):365-74. doi: 10.1002/gepi.20028. Genet Epidemiol. 2004. PMID: 15372618 Review.
-
Evaluating associations of haplotypes with traits.Genet Epidemiol. 2004 Dec;27(4):348-64. doi: 10.1002/gepi.20037. Genet Epidemiol. 2004. PMID: 15543638 Review.
Cited by
-
Bayesian survival analysis in genetic association studies.Bioinformatics. 2008 Sep 15;24(18):2030-6. doi: 10.1093/bioinformatics/btn351. Epub 2008 Jul 9. Bioinformatics. 2008. PMID: 18617538 Free PMC article.
-
Single Marker and Haplotype-Based Association Analysis of Semolina and Pasta Colour in Elite Durum Wheat Breeding Lines Using a High-Density Consensus Map.PLoS One. 2017 Jan 30;12(1):e0170941. doi: 10.1371/journal.pone.0170941. eCollection 2017. PLoS One. 2017. PMID: 28135299 Free PMC article.
-
Efficient whole-genome association mapping using local phylogenies for unphased genotype data.Bioinformatics. 2008 Oct 1;24(19):2215-21. doi: 10.1093/bioinformatics/btn406. Epub 2008 Jul 30. Bioinformatics. 2008. PMID: 18667442 Free PMC article.
-
Bayesian quantitative trait locus mapping using inferred haplotypes.Genetics. 2010 Mar;184(3):839-52. doi: 10.1534/genetics.109.113183. Epub 2010 Jan 4. Genetics. 2010. PMID: 20048050 Free PMC article.
-
New Genetic Approaches to AD: Lessons from APOE-TOMM40 Phylogenetics.Curr Neurol Neurosci Rep. 2016 May;16(5):48. doi: 10.1007/s11910-016-0643-8. Curr Neurol Neurosci Rep. 2016. PMID: 27039903 Review.
References
-
- Daly M, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–232. - PubMed
-
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. - PubMed
-
- Reich DE, Schaffner SF, Daly MJ, McVean G, Mullikin JC, et al. Human genome sequence variation and the influence of gene history, mutation and recombination. Nat Genet. 2002;32:135–142. - PubMed
-
- Molitor J, Marjoram P, Thomas D. Application of Bayesian spatial statistical methods to analysis of haplotypes effects and gene mapping. Genet Epidemiol. 2003;25:95–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
