

Cysteine-cysteine contact preference leads to target-focusing in protein folding
- PMID: 17617551
- PMCID: PMC1913149
- DOI: 10.1529/biophysj.106.097824
Cysteine-cysteine contact preference leads to target-focusing in protein folding
Abstract
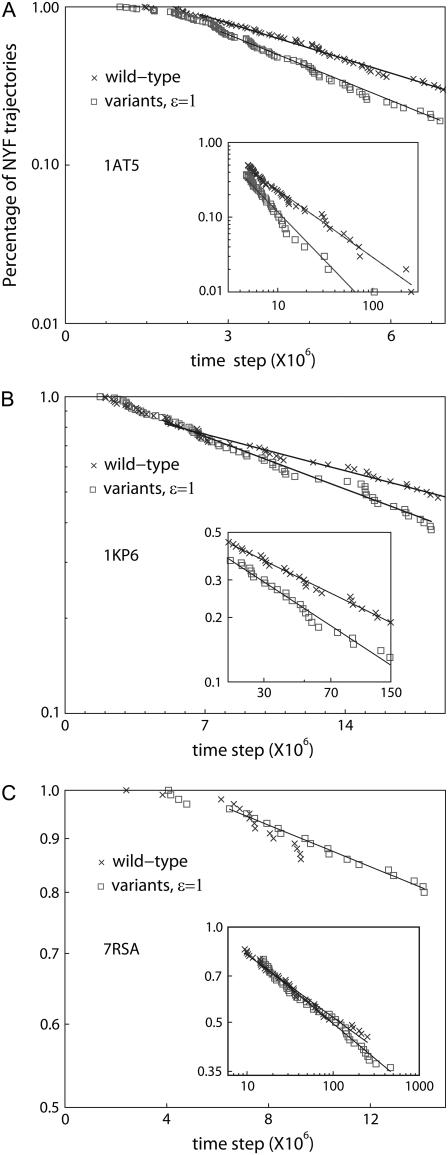
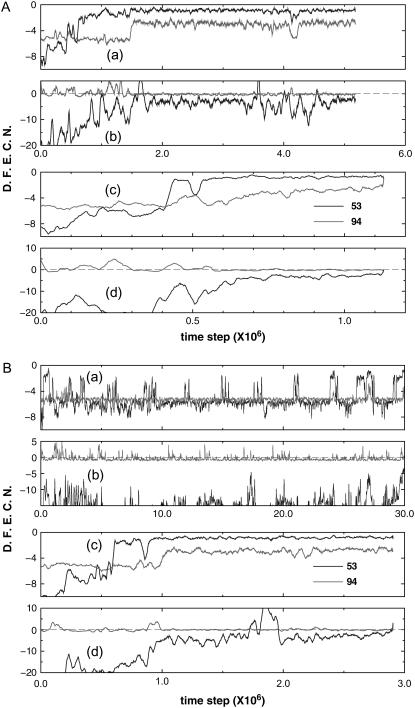
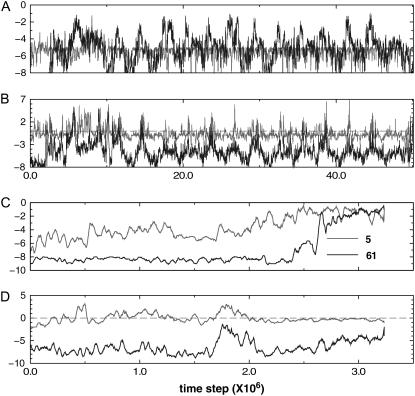
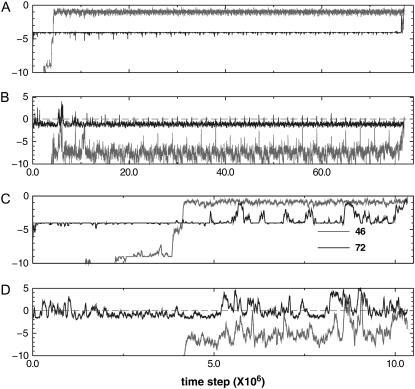
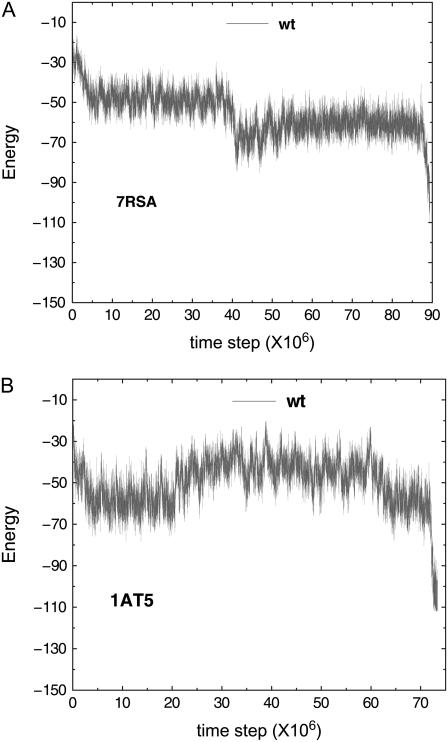
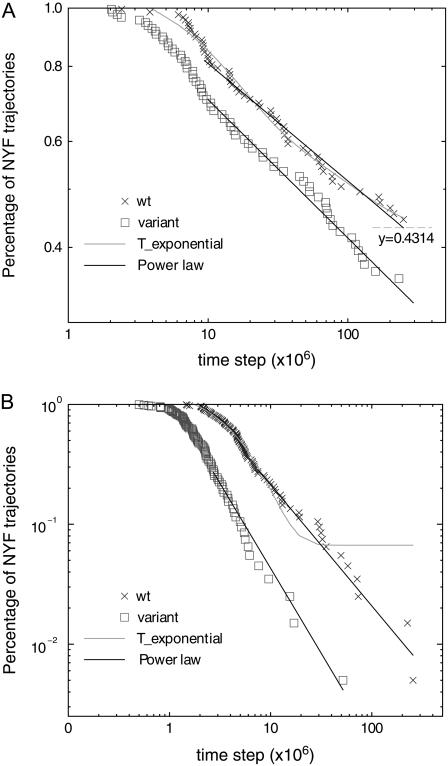
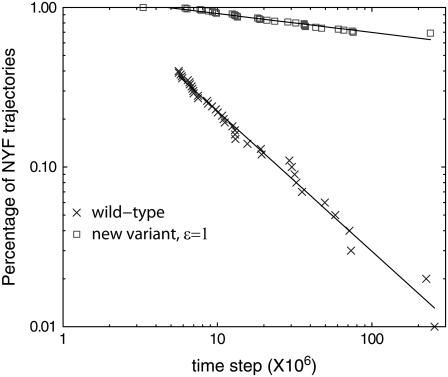
We perform a statistical analysis of amino-acid contacts to investigate possible preferences of amino-acid interactions. We include in the analysis only tertiary contacts, because they are less constrained--compared to secondary contacts--by proteins' backbone rigidity. Using proteins from the protein data bank, our analysis reveals an unusually high frequency of cysteine pairings relative to that expected from random. To elucidate the possible effects of cysteine interactions in folding, we perform molecular simulations on three cysteine-rich proteins. In particular, we investigate the difference in folding dynamics between a Gō-like model (where attraction only occurs between amino acids forming a native contact) and a variant model (where attraction between any two cysteines is introduced to mimic the formation/dissociation of native/nonnative disulfide bonds). We find that when attraction among cysteines is nonspecific and comparable to a solvent-averaged interaction, they produce a target-focusing effect that expedites folding of cysteine-rich proteins as a result of a reduction of conformational search space. In addition, the target-focusing effect also helps reduce glassiness by lowering activation energy barriers and kinetic frustration in the system. The concept of target-focusing also provides a qualitative understanding of a correlation between the rates of protein folding and parameters such as contact order and total contact distance.
Figures
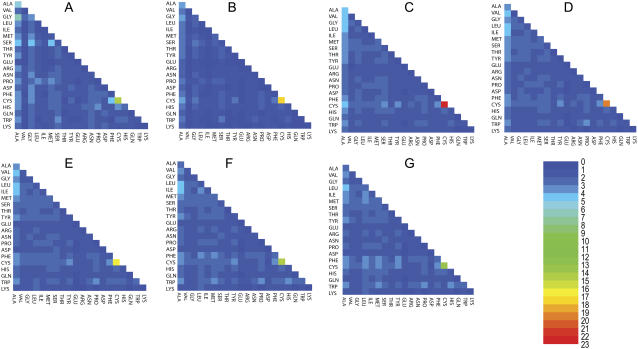
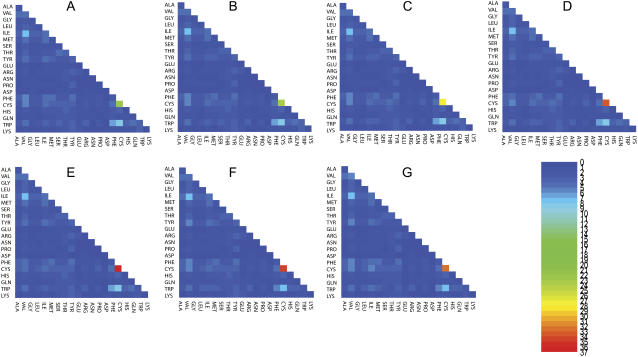
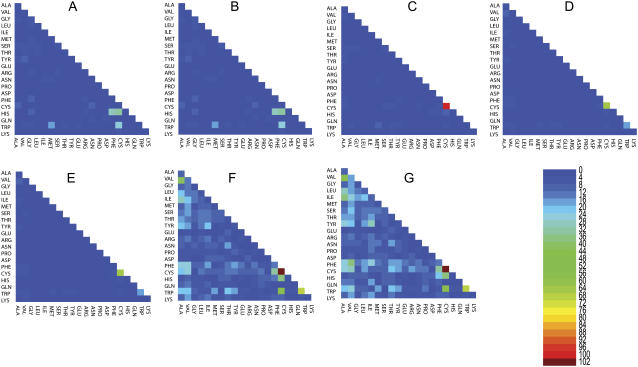
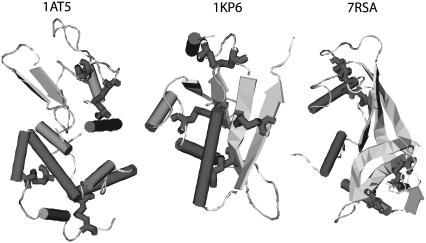
References
-
- Fersht, A. R. 1999. Structure and Mechanism in Protein Science. W. H. Freeman and Company, New York.
-
- Creighton, T. E. 1992. Protein Folding. W. H. Freeman and Company, New York.
-
- Miyazawa, S., and R. L. Jernigan. 1996. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 256:623–644. - PubMed
-
- Abkevich, V. I., and E. I. Shakhnovich. 2000. What can disulfide bonds tell us about protein energetics, function and folding: simulations and bioinformatics analysis. J. Mol. Biol. 300:975–985. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
