

BUB1 mediation of caspase-independent mitotic death determines cell fate
- PMID: 17620410
- PMCID: PMC2064447
- DOI: 10.1083/jcb.200702134
BUB1 mediation of caspase-independent mitotic death determines cell fate
Abstract
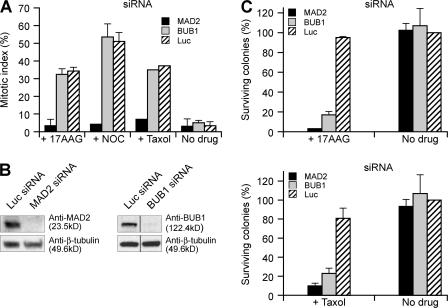
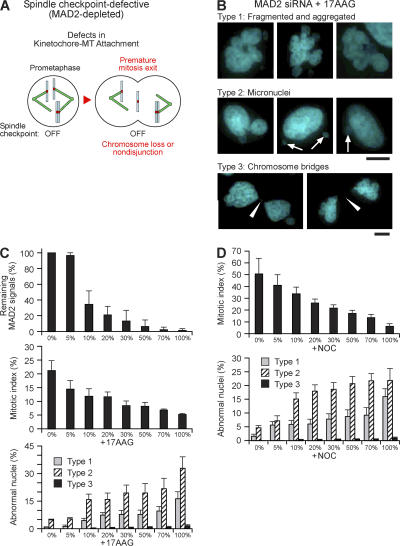
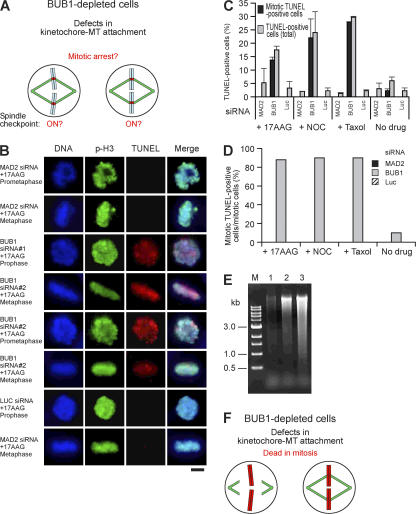
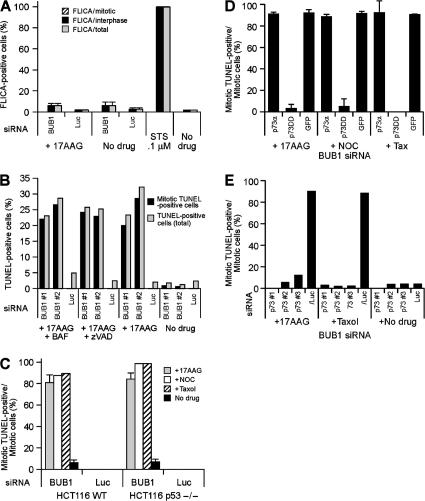
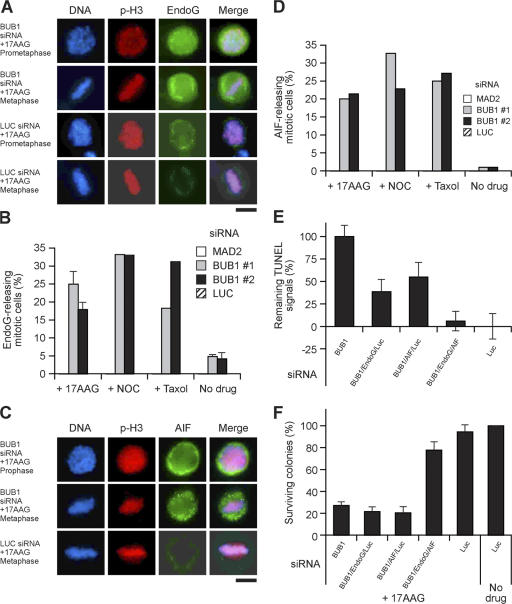
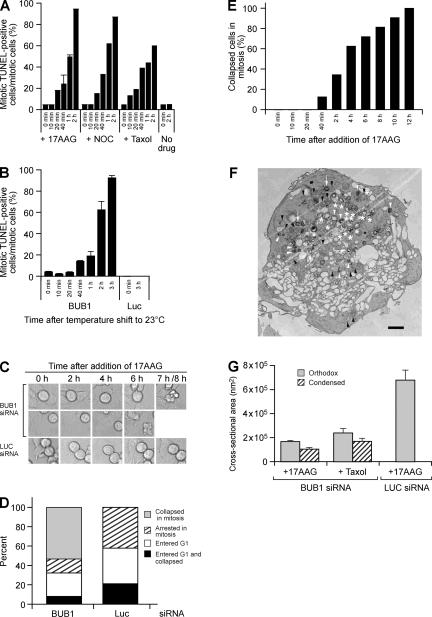
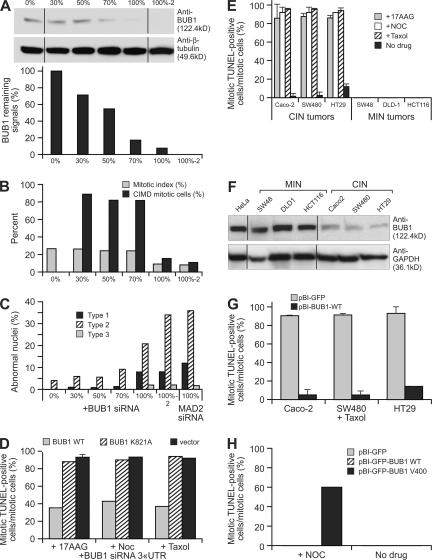
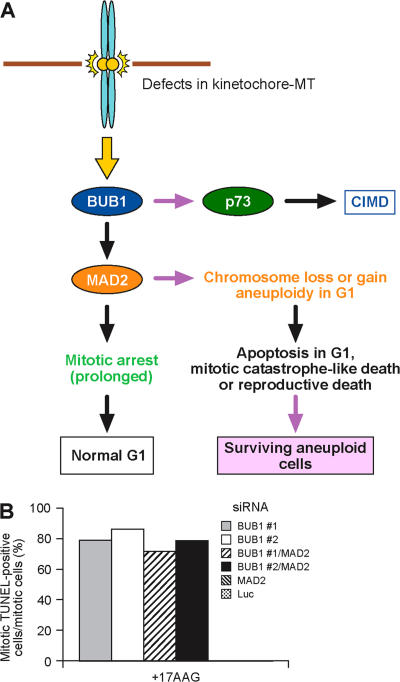
The spindle checkpoint that monitors kinetochore-microtubule attachment has been implicated in tumorigenesis; however, the relation between the spindle checkpoint and cell death remains obscure. In BUB1-deficient (but not MAD2-deficient) cells, conditions that activate the spindle checkpoint (i.e., cold shock or treatment with nocodazole, paclitaxel, or 17-AAG) induced DNA fragmentation during early mitosis. This mitotic cell death was independent of caspase activation; therefore, we named it caspase-independent mitotic death (CIMD). CIMD depends on p73, a homologue of p53, but not on p53. CIMD also depends on apoptosis-inducing factor and endonuclease G, which are effectors of caspase-independent cell death. Treatment with nocodazole, paclitaxel, or 17-AAG induced CIMD in cell lines derived from colon tumors with chromosome instability, but not in cells from colon tumors with microsatellite instability. This result was due to low BUB1 expression in the former cell lines. When BUB1 is completely depleted, aneuploidy rather than CIMD occurs. These results suggest that cells prone to substantial chromosome missegregation might be eliminated via CIMD.
Figures
Similar articles
-
Caspase-independent mitotic death (CIMD).Cell Cycle. 2008 Apr 15;7(8):1001-5. doi: 10.4161/cc.7.8.5720. Epub 2008 Feb 11. Cell Cycle. 2008. PMID: 18414023 Review.
-
BUB3 that dissociates from BUB1 activates caspase-independent mitotic death (CIMD).Cell Death Differ. 2010 Jun;17(6):1011-24. doi: 10.1038/cdd.2009.207. Epub 2010 Jan 8. Cell Death Differ. 2010. PMID: 20057499 Free PMC article.
-
Bub1 and aurora B cooperate to maintain BubR1-mediated inhibition of APC/CCdc20.J Cell Sci. 2005 Aug 15;118(Pt 16):3639-52. doi: 10.1242/jcs.02487. Epub 2005 Jul 26. J Cell Sci. 2005. PMID: 16046481
-
Induction of apoptosis by monastrol, an inhibitor of the mitotic kinesin Eg5, is independent of the spindle checkpoint.Mol Cancer Ther. 2006 Oct;5(10):2580-91. doi: 10.1158/1535-7163.MCT-06-0201. Mol Cancer Ther. 2006. PMID: 17041103
-
An essential role for p73 in regulating mitotic cell death.Cell Death Differ. 2010 May;17(5):787-800. doi: 10.1038/cdd.2009.181. Epub 2009 Dec 11. Cell Death Differ. 2010. PMID: 20010785
Cited by
-
BUB1 and SURVIVIN proteins are not degraded after a prolonged mitosis and accumulate in the nuclei of HCT116 cells.Cell Death Discov. 2016 Oct 24;2:16079. doi: 10.1038/cddiscovery.2016.79. eCollection 2016. Cell Death Discov. 2016. PMID: 27818790 Free PMC article.
-
Functional Significance of Aurora Kinases-p53 Protein Family Interactions in Cancer.Front Oncol. 2016 Nov 25;6:247. doi: 10.3389/fonc.2016.00247. eCollection 2016. Front Oncol. 2016. PMID: 27933271 Free PMC article. Review.
-
The Interaction of the Metallo-Glycopeptide Anti-Tumour Drug Bleomycin with DNA.Int J Mol Sci. 2018 May 4;19(5):1372. doi: 10.3390/ijms19051372. Int J Mol Sci. 2018. PMID: 29734689 Free PMC article. Review.
-
Kinases Involved in Both Autophagy and Mitosis.Int J Mol Sci. 2017 Aug 31;18(9):1884. doi: 10.3390/ijms18091884. Int J Mol Sci. 2017. PMID: 28858266 Free PMC article. Review.
-
SGT1-HSP90 complex is required for CENP-A deposition at centromeres.Cell Cycle. 2017 Sep 17;16(18):1683-1694. doi: 10.1080/15384101.2017.1325039. Epub 2017 Aug 17. Cell Cycle. 2017. PMID: 28816574 Free PMC article.
References
-
- Asnaghi, L., A. Calastretti, A. Bevilacqua, I. D'Agnano, G. Gatti, G. Canti, D. Delia, S. Capaccioli, and A. Nicolin. 2004. Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene. 23:5781–5791. - PubMed
-
- Banerji, U., A. O'Donnell, M. Scurr, S. Pacey, S. Stapleton, Y. Asad, L. Simmons, A. Maloney, F. Raynaud, M. Campbell, et al. 2005. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 23:4152–4161. - PubMed
-
- Barni, S., L. Sciola, A. Spano, and P. Pippia. 1996. Static cytofluorometry and fluorescence morphology of mitochondria and DNA in proliferating fibroblasts. Biotech. Histochem. 71:66–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
