

Protein folding by zipping and assembly
- PMID: 17620603
- PMCID: PMC1924571
- DOI: 10.1073/pnas.0703700104
Protein folding by zipping and assembly
Abstract
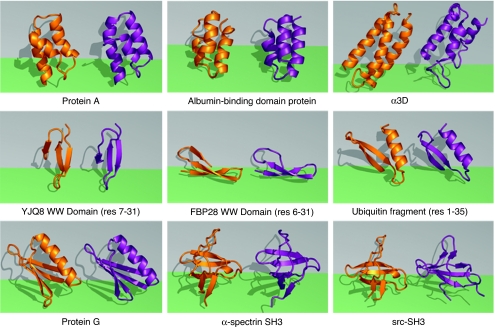
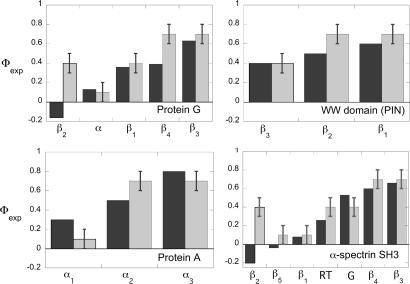
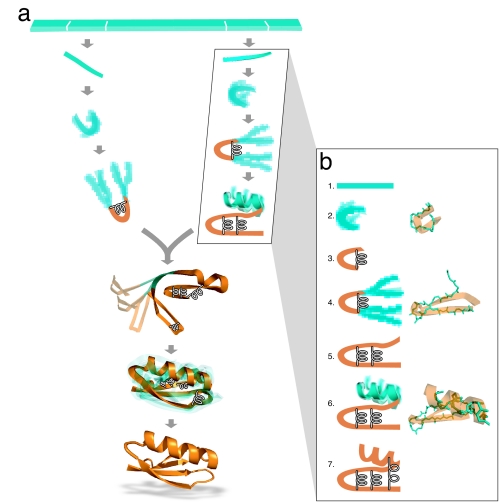
How do proteins fold so quickly? Some denatured proteins fold to their native structures in only microseconds, on average, implying that there is a folding "mechanism," i.e., a particular set of events by which the protein short-circuits a broader conformational search. Predicting protein structures using atomically detailed physical models is currently challenging. The most definitive proof of a putative folding mechanism would be whether it speeds up protein structure prediction in physical models. In the zipping and assembly (ZA) mechanism, local structuring happens first at independent sites along the chain, then those structures either grow (zip) or coalescence (assemble) with other structures. Here, we apply the ZA search mechanism to protein native structure prediction by using the AMBER96 force field with a generalized Born/surface area implicit solvent model and sampling by replica exchange molecular dynamics. Starting from open denatured conformations, our algorithm, called the ZA method, converges to an average of 2.2 A from the Protein Data Bank native structures of eight of nine proteins that we tested, which ranged from 25 to 73 aa in length. In addition, experimental Phi values, where available on these proteins, are consistent with the predicted routes. We conclude that ZA is a viable model for how proteins physically fold. The present work also shows that physics-based force fields are quite good and that physics-based protein structure prediction may be practical, at least for some small proteins.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Blind test of physics-based prediction of protein structures.Biophys J. 2009 Feb;96(3):917-24. doi: 10.1016/j.bpj.2008.11.009. Biophys J. 2009. PMID: 19186130 Free PMC article.
-
Exploring zipping and assembly as a protein folding principle.Proteins. 2007 Mar 1;66(4):877-88. doi: 10.1002/prot.21234. Proteins. 2007. PMID: 17154424
-
Free energy landscape of protein folding in water: explicit vs. implicit solvent.Proteins. 2003 Nov 1;53(2):148-61. doi: 10.1002/prot.10483. Proteins. 2003. PMID: 14517967
-
Protein-folding dynamics: overview of molecular simulation techniques.Annu Rev Phys Chem. 2007;58:57-83. doi: 10.1146/annurev.physchem.58.032806.104614. Annu Rev Phys Chem. 2007. PMID: 17034338 Review.
-
Intrinsically disordered proteins in a physics-based world.Int J Mol Sci. 2010;11(12):5292-309. doi: 10.3390/ijms11125292. Epub 2010 Dec 21. Int J Mol Sci. 2010. PMID: 21614208 Free PMC article. Review.
Cited by
-
Interdiction in the Early Folding of the p53 DNA-Binding Domain Leads to Its Amyloid-Like Misfolding.Molecules. 2022 Jul 27;27(15):4810. doi: 10.3390/molecules27154810. Molecules. 2022. PMID: 35956758 Free PMC article.
-
Mimicking the folding pathway to improve homology-free protein structure prediction.Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):3734-9. doi: 10.1073/pnas.0811363106. Epub 2009 Feb 23. Proc Natl Acad Sci U S A. 2009. PMID: 19237560 Free PMC article.
-
ASTRO-FOLD 2.0: an Enhanced Framework for Protein Structure Prediction.AIChE J. 2012 May 1;58(5):1619-1637. doi: 10.1002/aic.12669. Epub 2011 May 31. AIChE J. 2012. PMID: 23049093 Free PMC article.
-
Advanced techniques for constrained internal coordinate molecular dynamics.J Comput Chem. 2013 Apr 30;34(11):904-14. doi: 10.1002/jcc.23200. Epub 2013 Jan 23. J Comput Chem. 2013. PMID: 23345138 Free PMC article.
-
PRIMO: A Transferable Coarse-grained Force Field for Proteins.J Chem Theory Comput. 2013 Aug 13;9(8):3769-3788. doi: 10.1021/ct400230y. J Chem Theory Comput. 2013. PMID: 23997693 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
