

The role of zinc in cerebral ischemia
- PMID: 17622314
- PMCID: PMC1952671
- DOI: 10.2119/2007–00044.Galasso
The role of zinc in cerebral ischemia
Abstract
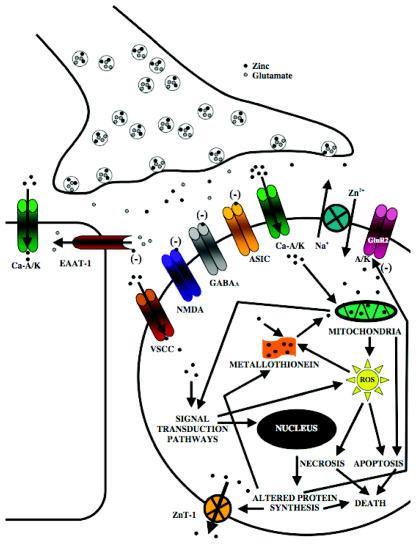
Ischemic stroke is one of the most pervasive life-threatening neurological conditions for which there currently exists limited therapeutic intervention beyond prevention. As calcium-focused neuroprotective strategies have met with limited clinical success, it is imperative that alternative therapeutic targets be considered in the attempt to antagonize ischemic-mediated injury. As such, zinc, which is able to function both as a signaling mediator and neurotoxin, has been implicated in cerebral ischemia. While zinc was first purported to have a role in cerebral ischemia nearly twenty years ago, our understanding of how zinc mediates ischemic injury is still in its relative infancy. Within this review, we examine some of the studies by which zinc has exerted either neuroprotective or neurotoxic effects during global and focal cerebral ischemia.
Figures
References
-
- Rothwell PM. The high cost of not funding stroke research: a comparison with heart disease and cancer. Lancet. 2001;357:1612–6. - PubMed
-
- Caplan LR. Caplan’s Stroke: A Clinical Approach. Butterworth-Heinemann; Boston, Massachusetts: 2000. 556.
-
- Bambauer KZ, Johnston SC, Bambauer DE, Zivin JA. Reasons why few patients with acute stroke receive tissue plasminogen activator. Arch Neurol. 2006;63:661–4. - PubMed
-
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–14. - PubMed
-
- Sensi SL, Jeng JM. Rethinking the excitotoxic ionic milieu: the emerging role of Zn(2+) in ischemic neuronal injury. Curr Mol Med. 2004;4:87–111. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous