

Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death
- PMID: 17622797
- PMCID: PMC2779565
- DOI: 10.4161/auto.4625
Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death
Abstract
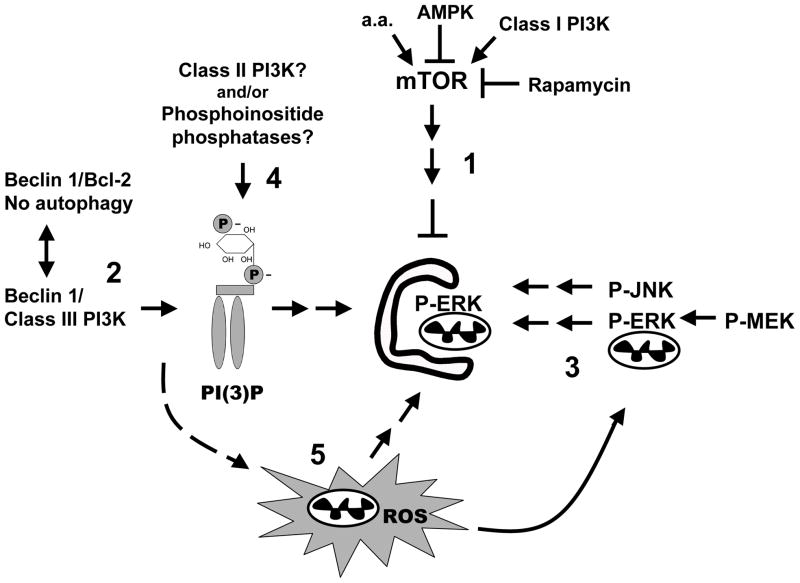
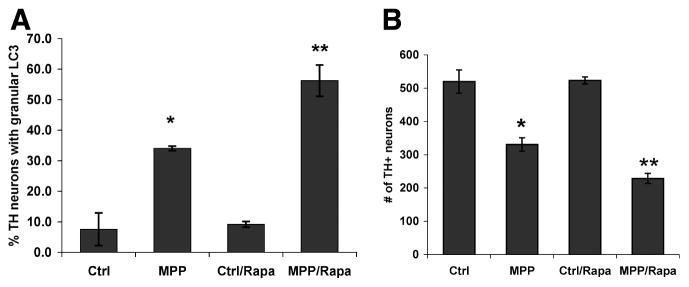
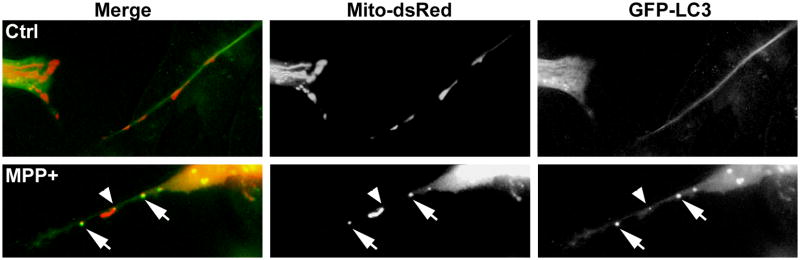
Growing evidence supports an active role for dysregulated macroautophagy (autophagic stress) in neuronal cell death and neurodegeneration. Alterations in mitochondrial function and dynamics are also strongly implicated in neurodegenerative diseases. Interestingly, whereas the core autophagy machinery is evolutionarily conserved and shared among constitutive and induced or selective autophagy, recent studies implicate distinct mechanisms regulating mitochondrial autophagy (mitophagy) in response to general autophagic stimuli. Little is known about pathways regulating selective, damage-induced mitophagy. We found that the parkinsonian neurotoxin MPP(+) induces autophagy and mitochondrial degradation that is inhibited by siRNA knockdown of autophagy proteins Atg5, Atg7 and Atg8, but occurs independently of Beclin 1, a component of the class III (PIK3C3/Vps34) phosphoinositide 3-kinase (PI3K) complex. Instead, MPP(+)-induced mitophagy is dependent upon MAPK signaling. Interestingly, all treatments that inhibited autophagy also conferred protection from MPP(+)-induced cell death. A prior human tissue study further supports a role for ERK/MAPK-regulated autophagy in Parkinson's and Lewy body diseases. As competition for limiting amounts of Beclin 1 may serve to prevent harmful overactivation of autophagy, understanding mechanisms that bypass or complement a requirement for PI3K-Beclin 1 activity could lead to strategies to modulate autophagic stress in injured or degenerating neurons.
Figures
References
-
- Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci. 2007;120:1707–16. - PubMed
-
- Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Jr, Davis RE, Parker WD., Jr Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol. 1996;40:663–71. - PubMed
-
- Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem. 2005;280:42026–35. - PubMed
-
- Abbott RD, Ross GW, White LR, Sanderson WT, Burchfiel CM, Kashon M, Sharp DS, Masaki KH, Curb JD, Petrovitch H. Environmental, life-style, and physical precursors of clinical Parkinson’s disease: recent findings from the Honolulu-Asia Aging Study. J Neurol. 2003;250(Suppl 3):III30–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous