

IP receptor-dependent activation of PPARgamma by stable prostacyclin analogues
- PMID: 17624303
- PMCID: PMC1997304
- DOI: 10.1016/j.bbrc.2007.06.135
IP receptor-dependent activation of PPARgamma by stable prostacyclin analogues
Abstract
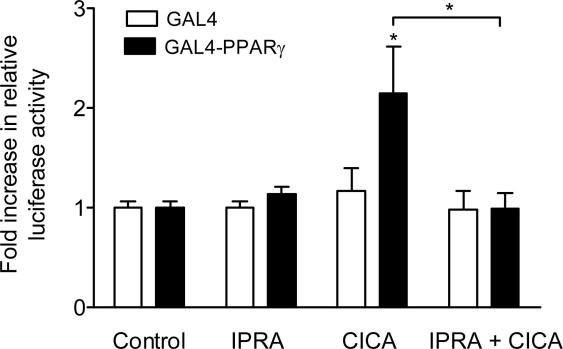
Stable prostacyclin analogues can signal through cell surface IP receptors or by ligand binding to nuclear peroxisome proliferator-activated receptors (PPARs). So far these agents have been reported to activate PPARalpha and PPARdelta but not PPARgamma. Given PPARgamma agonists and prostacyclin analogues both inhibit cell proliferation, we postulated that the IP receptor might elicit PPARgamma activation. Using a dual luciferase reporter gene assay in HEK-293 cells stably expressing the IP receptor or empty vector, we found that prostacyclin analogues only activated PPARgamma in the presence of the IP receptor. Moreover, the novel IP receptor antagonist, RO1138452, but not inhibitors of the cyclic AMP pathway, prevented activation. Likewise, the anti-proliferative effects of treprostinil observed in IP receptor expressing cells, were partially inhibited by the PPARgamma antagonist, GW9662. We conclude that PPARgamma is activated through the IP receptor via a cyclic AMP-independent mechanism and contributes to the anti-growth effects of prostacyclin analogues.
Figures





Similar articles
-
Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension.Am J Respir Crit Care Med. 2010 Nov 1;182(9):1161-70. doi: 10.1164/rccm.201001-0011OC. Epub 2010 Jul 9. Am J Respir Crit Care Med. 2010. PMID: 20622039 Free PMC article.
-
Role of prostacyclin versus peroxisome proliferator-activated receptor beta receptors in prostacyclin sensing by lung fibroblasts.Am J Respir Cell Mol Biol. 2006 Feb;34(2):242-6. doi: 10.1165/rcmb.2005-0289OC. Epub 2005 Oct 20. Am J Respir Cell Mol Biol. 2006. PMID: 16239641
-
Prostacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator--activated receptor gamma.Cancer Prev Res (Phila). 2008 Oct;1(5):349-56. doi: 10.1158/1940-6207.CAPR-08-0145. Cancer Prev Res (Phila). 2008. PMID: 19138979 Free PMC article.
-
The mechanistic basis of prostacyclin and its stable analogues in pulmonary arterial hypertension: Role of membrane versus nuclear receptors.Prostaglandins Other Lipid Mediat. 2015 Jul;120:56-71. doi: 10.1016/j.prostaglandins.2015.04.007. Epub 2015 Apr 23. Prostaglandins Other Lipid Mediat. 2015. PMID: 25917921 Review.
-
A novel pathway of prostacyclin signaling-hanging out with nuclear receptors.Endocrinology. 2002 Sep;143(9):3207-10. doi: 10.1210/en.2002-220159. Endocrinology. 2002. PMID: 12193530 Review.
Cited by
-
Oral treprostinil improves pulmonary vascular compliance in pulmonary arterial hypertension.Respir Med. 2022 Mar;193:106744. doi: 10.1016/j.rmed.2022.106744. Epub 2022 Jan 19. Respir Med. 2022. PMID: 35134631 Free PMC article. Clinical Trial.
-
Current Overview of the Biology and Pharmacology in Sugen/Hypoxia-Induced Pulmonary Hypertension in Rats.J Aerosol Med Pulm Drug Deliv. 2024 Oct;37(5):241-283. doi: 10.1089/jamp.2024.0016. J Aerosol Med Pulm Drug Deliv. 2024. PMID: 39388691 Review.
-
PPARδ activity in cardiovascular diseases: A potential pharmacological target.PPAR Res. 2009;2009:745821. doi: 10.1155/2009/745821. Epub 2009 Mar 23. PPAR Res. 2009. PMID: 19325917 Free PMC article.
-
Stimulation of fat storage by prostacyclin and selective agonists of prostanoid IP receptor during the maturation phase of cultured adipocytes.Cytotechnology. 2016 Dec;68(6):2417-2429. doi: 10.1007/s10616-016-9960-7. Epub 2016 Mar 5. Cytotechnology. 2016. PMID: 26946143 Free PMC article.
-
Endogenous synthesis of prostacyclin was positively regulated during the maturation phase of cultured adipocytes.Cytotechnology. 2014 Aug;66(4):635-46. doi: 10.1007/s10616-013-9616-9. Epub 2013 Jul 25. Cytotechnology. 2014. PMID: 23884720 Free PMC article.
References
-
- Narumiya S., Sugimoto Y., Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol. Rev. 1999;79:1193–1226. - PubMed
-
- Vane J., Corin R.E. Prostacyclin: a vascular mediator. Eur. J. Vasc. Endovasc. Surg. 2003;26:571–578. - PubMed
-
- Wise H. Multiple signalling options for prostacyclin. Acta Pharmacol. Sin. 2003;24:625–630. - PubMed
-
- Lim H., Dey S.K. A novel pathway of prostacyclin signaling-hanging out with nuclear receptors. Endocrinology. 2002;143:3207–3210. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
