

Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence
- PMID: 17626202
- PMCID: PMC6672614
- DOI: 10.1523/JNEUROSCI.4540-06.2007
Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence
Abstract
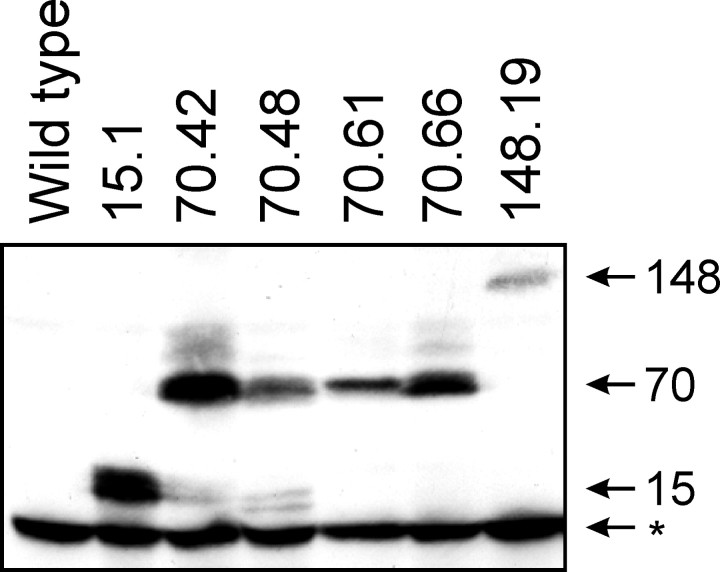
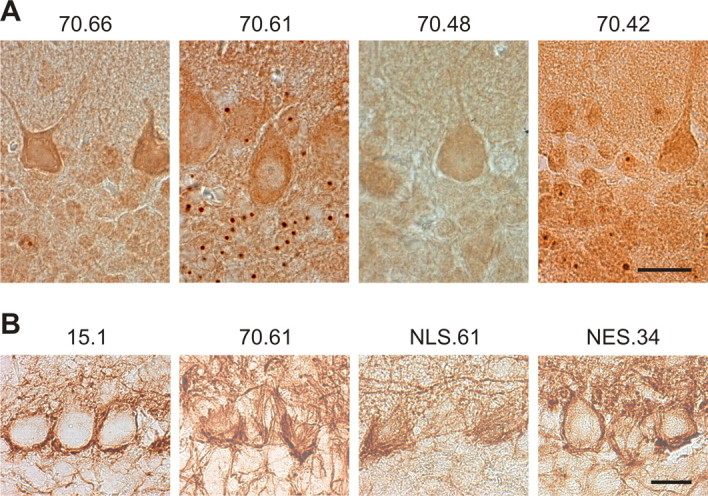
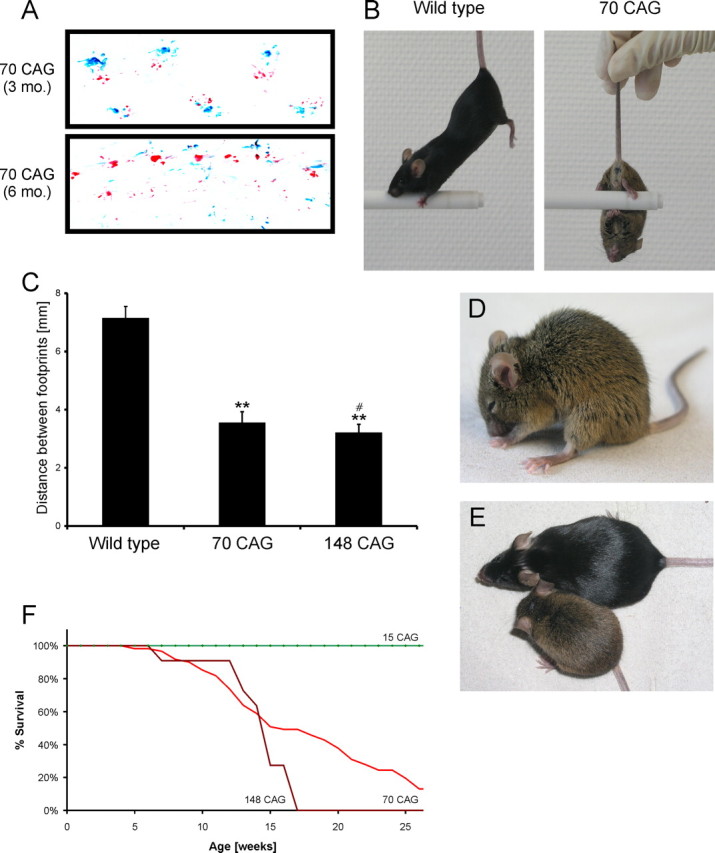
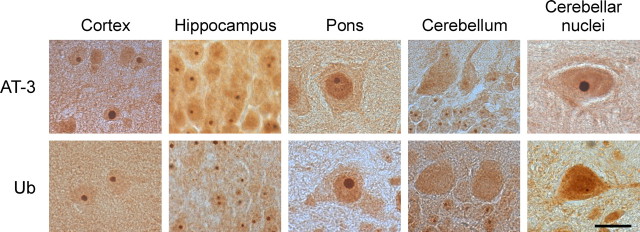
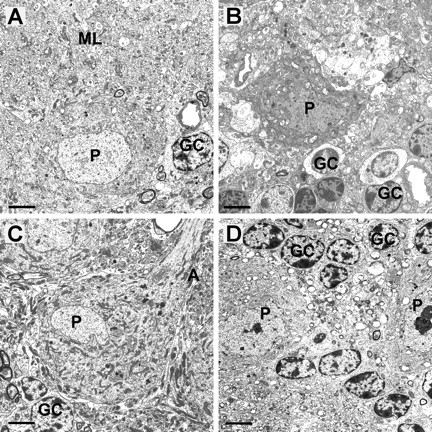
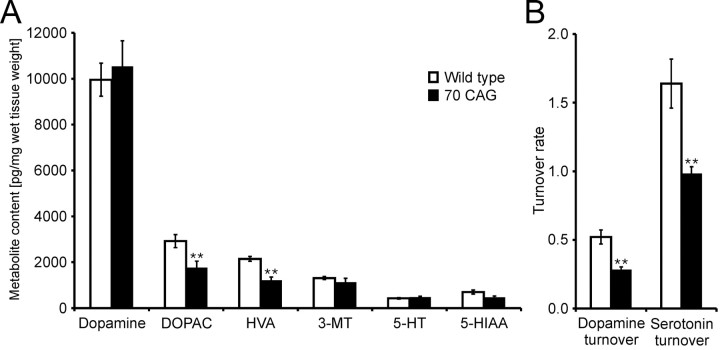
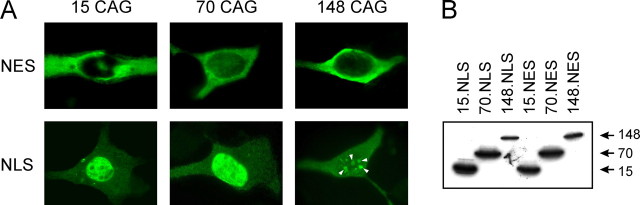
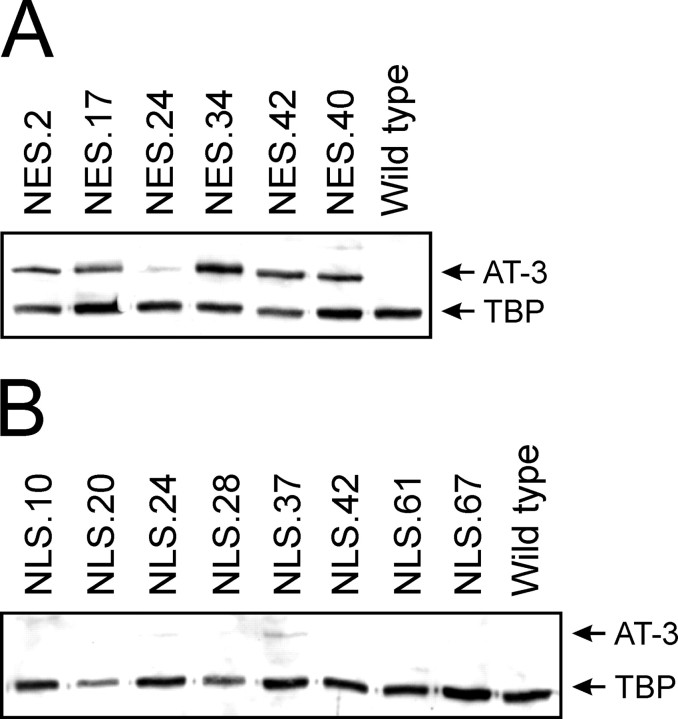
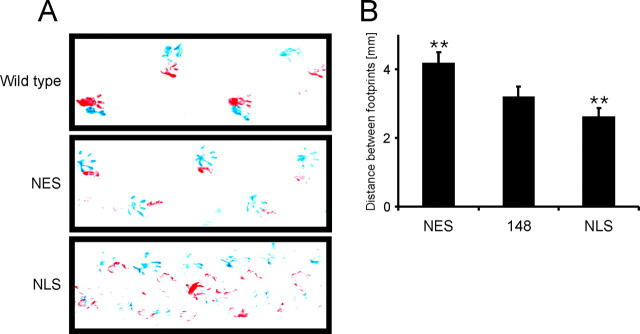
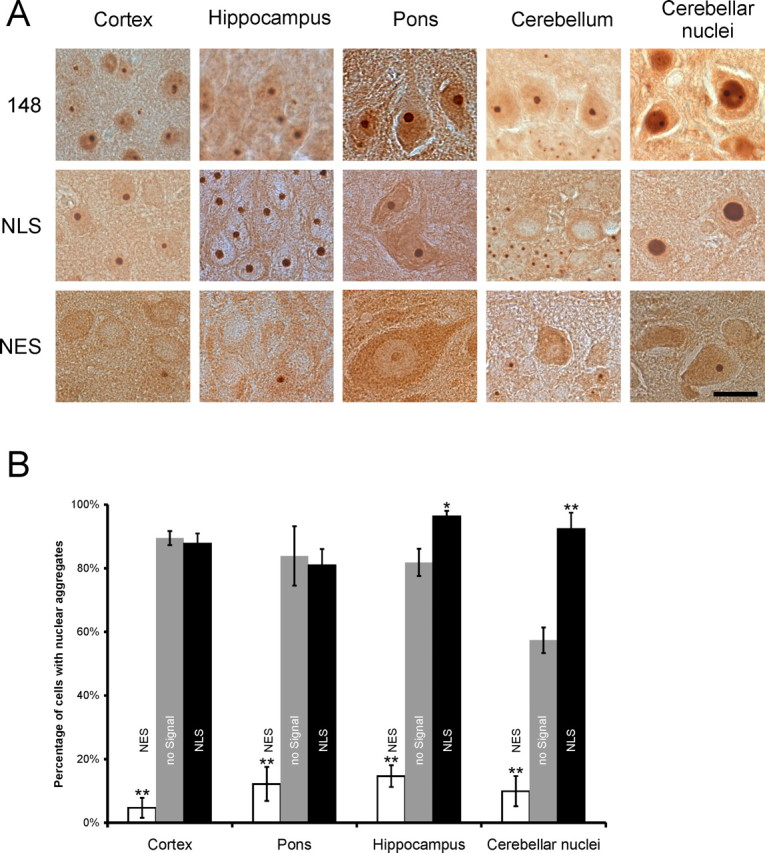
Spinocerebellar ataxia type 3 (SCA3) is an autosomal dominantly inherited neurodegenerative disorder caused by the expansion of a CAG repeat in the MJD1 gene resulting in an expanded polyglutamine repeat in the ataxin-3 protein. To study the course of the disease, we generated transgenic mice for SCA3 using full-length ataxin-3 constructs containing 15, 70, or 148 CAG repeats, respectively. Control mice (15 CAGs) were phenotypically normal and had no neuropathological findings. However, mice transgenic for ataxin-3 with expanded polyglutamine repeats were severely affected by a strong neurological phenotype with tremor, behavioral deficits, strongly reduced motor and exploratory activity, a hunchback, and premature death at 3 to 6 months of age. Neuropathological examination by immunohistochemical staining revealed ubiquitin- and ataxin-3-positive intranuclear inclusion bodies in a multitude of neurons. Directing ataxin-3 with 148 CAGs to the nucleus revealed an even more pronounced phenotype with more inclusions and earlier death, whereas mice transgenic with the same construct but attached to a nuclear export signal developed a milder phenotype with less inclusions. These studies indicate that nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3 in vivo.
Figures
References
-
- Albrecht M, Golatta M, Wüllner U, Lengauer T. Structural and functional analysis of ataxin-2 and ataxin-3. Eur J Biochem. 2004;271:3155–3170. - PubMed
-
- Baybutt H, Manson J. Characterisation of two promoters for prion protein (PrP) gene expression in neuronal cells. Gene. 1997;184:125–131. - PubMed
-
- Benn CL, Landles C, Li H, Strand AD, Woodman B, Sathasivam K, Li SH, Ghazi-Noori S, Hockly E, Faruque SM, Cha JH, Sharpe PT, Olson JM, Li XJ, Bates GP. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington's disease. Hum Mol Genet. 2005;14:3065–3078. - PubMed
-
- Bjorkqvist M, Fex M, Renstrom E, Wierup N, Petersen A, Gil J, Bacos K, Popovic N, Li JY, Sundler F, Brundin P, Mulder H. The R6/2 transgenic mouse model of Huntington's disease develops diabetes due to deficient beta-cell mass and exocytosis. Hum Mol Genet. 2005;14:565–574. - PubMed
-
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases