

RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation
- PMID: 17626792
- PMCID: PMC1920174
- DOI: 10.1101/gad.1544107
RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation
Abstract
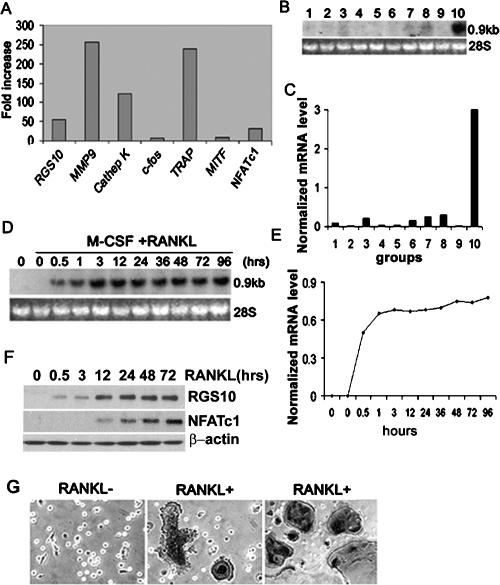
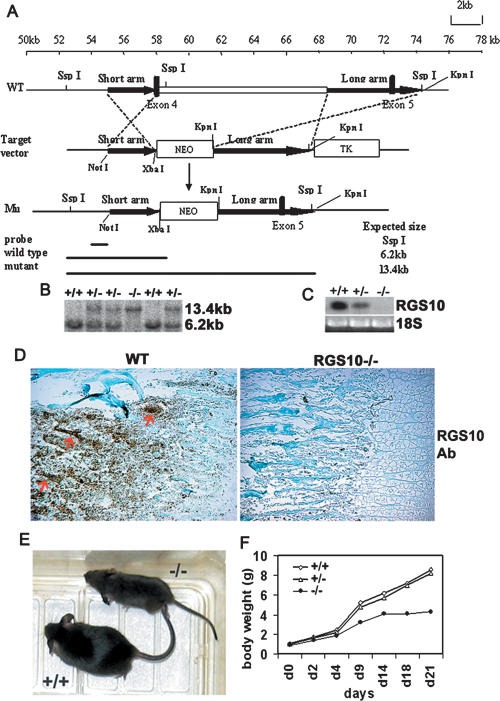
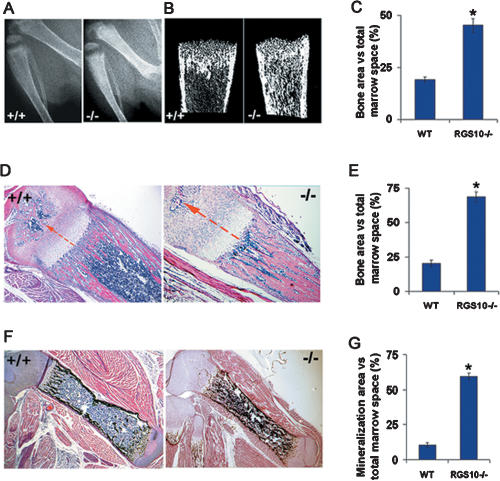
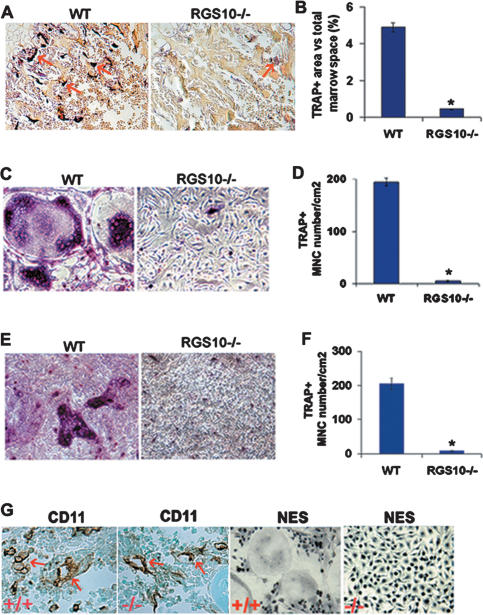
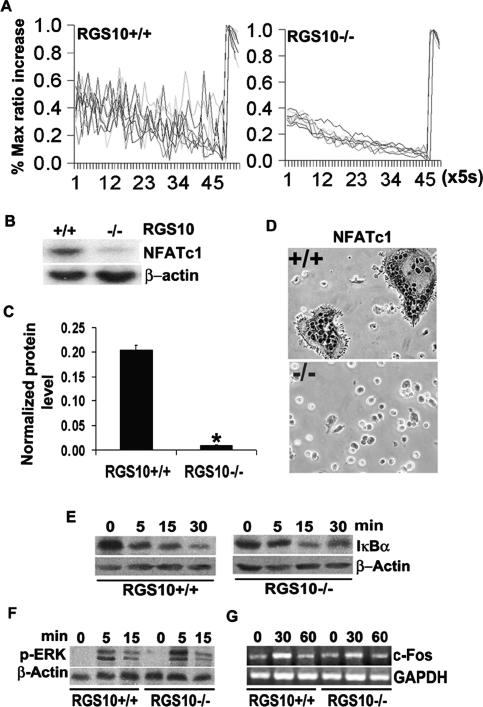
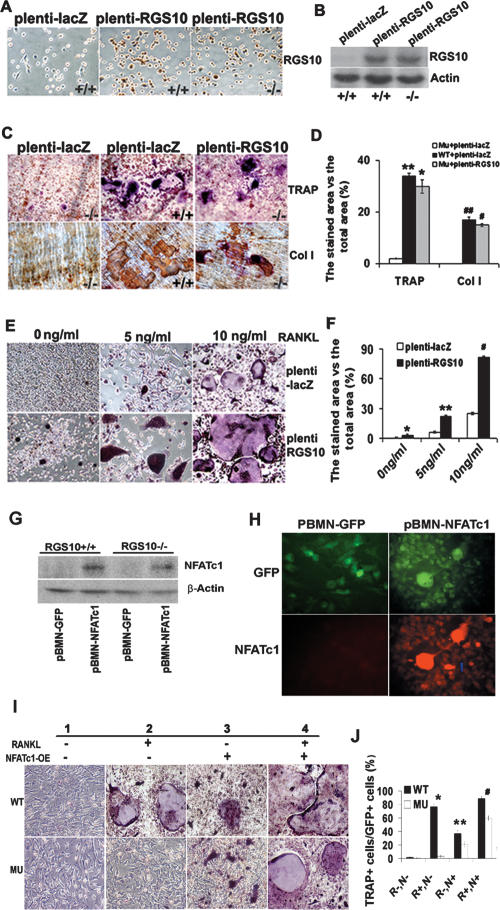
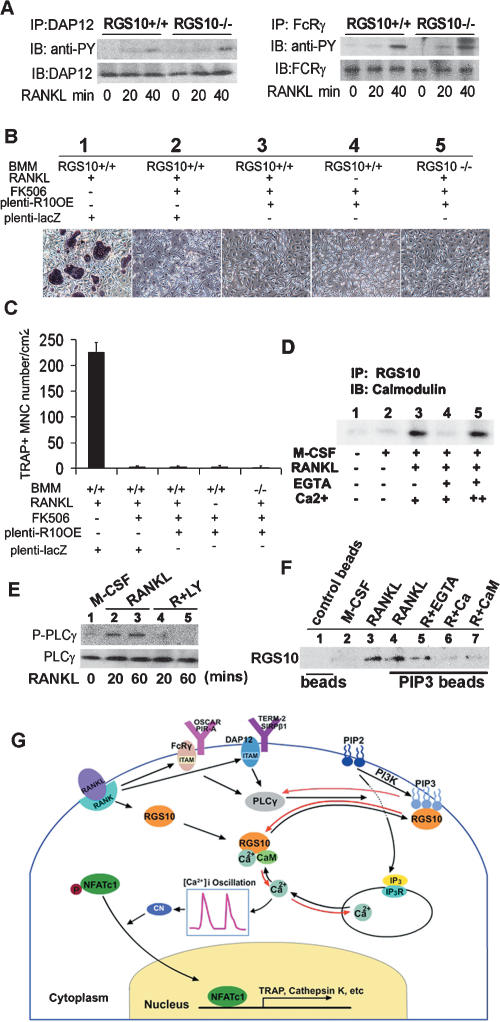
Increased osteoclastic resorption leads to many bone diseases, including osteoporosis and rheumatoid arthritis. While rapid progress has been made in characterizing osteoclast differentiation signaling pathways, how receptor activator of nuclear factor kappaB (NF-kappaB) ligand (RANKL) evokes essential [Ca2+]i oscillation signaling remains unknown. Here, we characterized RANKL-induced signaling proteins and found regulator of G-protein signaling 10 (RGS10) is predominantly expressed in osteoclasts. We generated RGS10-deficient (RGS10-/-) mice that exhibited severe osteopetrosis and impaired osteoclast differentiation. Our data demonstrated that ectopic expression of RGS10 dramatically increased the sensitivity of osteoclast differentiation to RANKL signaling; the deficiency of RGS10 resulted in the absence of [Ca2+]i oscillations and loss of NFATc1; ectopic NFATc1 expression rescues impaired osteoclast differentiation from deletion of RGS10; phosphatidylinositol 3,4,5-trisphosphate (PIP3) is essential to PLCgamma activation; and RGS10 competitively interacts with Ca2+/calmodulin and PIP3 in a [Ca2+]i-dependent manner to mediate PLCgamma activation and [Ca2+]i oscillations. Our results revealed a mechanism through which RGS10 specifically regulates the RANKL-evoked RGS10/calmodulin-[Ca2+]i oscillation-calcineurin-NFATc1 signaling pathway in osteoclast differentiation using an in vivo model. RGS10 provides a potential therapeutic target for the treatment of bone diseases.
Figures
References
-
- Abramow-Newerly M., Roy A.A., Nunn C., Chidiac P., Roy A.A., Nunn C., Chidiac P., Nunn C., Chidiac P., Chidiac P. RGS proteins have a signalling complex: Interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell. Signal. 2006;18:579–591. - PubMed
-
- Appleton C.T., James C.G., Beier F., James C.G., Beier F., Beier F. Regulator of G-protein signaling (RGS) proteins differentially control chondrocyte differentiation. J. Cell. Physiol. 2006;207:735–745. - PubMed
-
- Baumeister W., Walz J., Zuhl F., Seemuller E., Walz J., Zuhl F., Seemuller E., Zuhl F., Seemuller E., Seemuller E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. - PubMed
-
- Boyle W.J., Simonet W.S., Lacey D.L., Simonet W.S., Lacey D.L., Lacey D.L. Osteoclast differentiation and activation. Nature. 2003;423:337–342. - PubMed
-
- Cunningham M.L., Waldo G.L., Hollinger S., Hepler J.R., Harden T.K., Waldo G.L., Hollinger S., Hepler J.R., Harden T.K., Hollinger S., Hepler J.R., Harden T.K., Hepler J.R., Harden T.K., Harden T.K. Protein kinase C phosphorylates RGS2 and modulates its capacity for negative regulation of Gα 11 signaling. J. Biol. Chem. 2001;276:5438–5444. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous