

Comparative genomic and protein sequence analyses of a complex system controlling bacterial chemotaxis
- PMID: 17628132
- PMCID: PMC2754700
- DOI: 10.1016/S0076-6879(06)22001-9
Comparative genomic and protein sequence analyses of a complex system controlling bacterial chemotaxis
Abstract
Molecular machinery governing bacterial chemotaxis consists of the CheA-CheY two-component system, an array of specialized chemoreceptors, and several auxiliary proteins. It has been studied extensively in Escherichia coli and, to a significantly lesser extent, in several other microbial species. Emerging evidence suggests that homologous signal transduction pathways regulate not only chemotaxis, but several other cellular functions in various bacterial species. The availability of genome sequence data for hundreds of organisms enables productive study of this system using comparative genomics and protein sequence analysis. This chapter describes advances in genomics of the chemotaxis signal transduction system, provides information on relevant bioinformatics tools and resources, and outlines approaches toward developing a computational framework for predicting important biological functions from raw genomic data based on available experimental evidence.
Figures
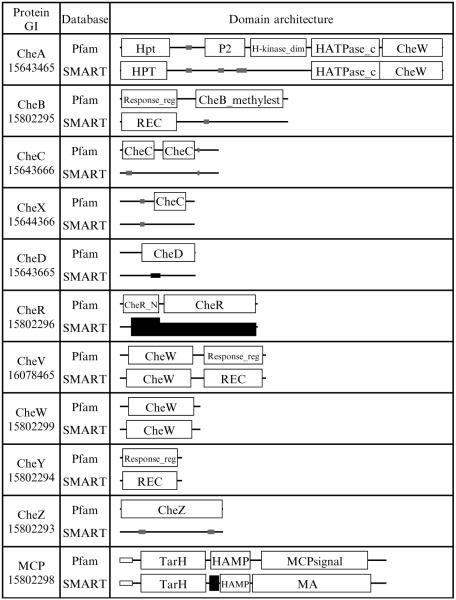
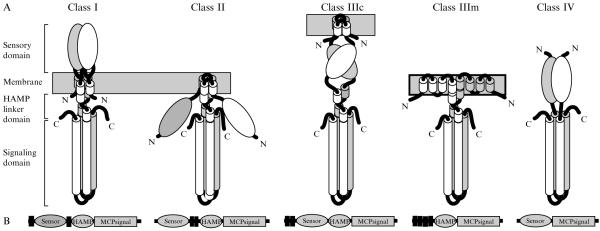
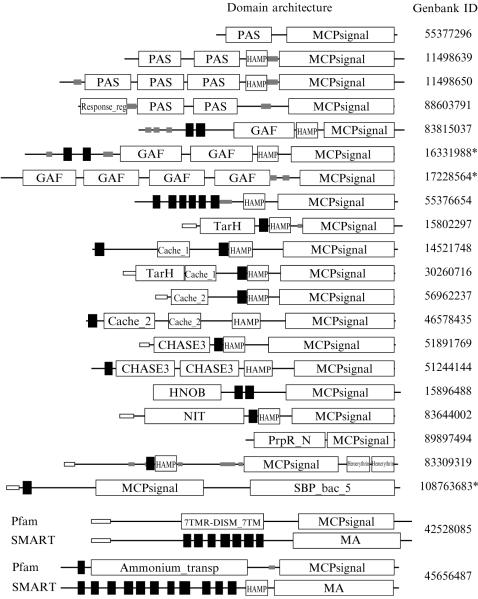
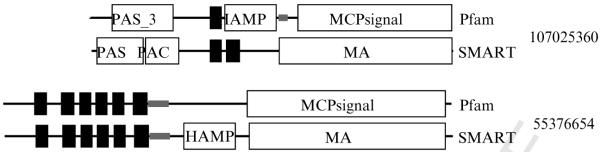
Similar articles
-
CheA, CheW, and CheY are required for chemotaxis to oxygen and sugars of the phosphotransferase system in Escherichia coli.J Bacteriol. 1995 Oct;177(20):6011-4. doi: 10.1128/jb.177.20.6011-6014.1995. J Bacteriol. 1995. PMID: 7592359 Free PMC article.
-
CobB regulates Escherichia coli chemotaxis by deacetylating the response regulator CheY.Mol Microbiol. 2010 Jun 1;76(5):1162-74. doi: 10.1111/j.1365-2958.2010.07125.x. Epub 2010 Mar 16. Mol Microbiol. 2010. PMID: 20345663 Free PMC article.
-
Determinants of chemotactic signal amplification in Escherichia coli.J Mol Biol. 2001 Mar 16;307(1):119-35. doi: 10.1006/jmbi.2000.4389. J Mol Biol. 2001. PMID: 11243808
-
Signaling and sensory adaptation in Escherichia coli chemoreceptors: 2015 update.Trends Microbiol. 2015 May;23(5):257-66. doi: 10.1016/j.tim.2015.03.003. Epub 2015 Mar 30. Trends Microbiol. 2015. PMID: 25834953 Free PMC article. Review.
-
Information processing in bacterial chemotaxis.Sci STKE. 2002 May 14;2002(132):pe25. doi: 10.1126/stke.2002.132.pe25. Sci STKE. 2002. PMID: 12011495 Review.
Cited by
-
Chemotaxis in Campylobacter jejuni.Eur J Microbiol Immunol (Bp). 2012 Mar;2(1):24-31. doi: 10.1556/EuJMI.2.2012.1.5. Epub 2012 Mar 17. Eur J Microbiol Immunol (Bp). 2012. PMID: 24611118 Free PMC article. Review.
-
Direct evidence that the carboxyl-terminal sequence of a bacterial chemoreceptor is an unstructured linker and enzyme tether.Protein Sci. 2011 Nov;20(11):1856-66. doi: 10.1002/pro.719. Epub 2011 Sep 15. Protein Sci. 2011. PMID: 21858888 Free PMC article.
-
Elements of the cellular metabolic structure.Front Mol Biosci. 2015 Apr 28;2:16. doi: 10.3389/fmolb.2015.00016. eCollection 2015. Front Mol Biosci. 2015. PMID: 25988183 Free PMC article.
-
Site-specific methylation in Bacillus subtilis chemotaxis: effect of covalent modifications to the chemotaxis receptor McpB.Microbiology (Reading). 2011 Jan;157(Pt 1):56-65. doi: 10.1099/mic.0.044685-0. Epub 2010 Sep 23. Microbiology (Reading). 2011. PMID: 20864474 Free PMC article.
-
Thriving in Wetlands: Ecophysiology of the Spiral-Shaped Methanotroph Methylospira mobilis as Revealed by the Complete Genome Sequence.Microorganisms. 2019 Dec 11;7(12):683. doi: 10.3390/microorganisms7120683. Microorganisms. 2019. PMID: 31835835 Free PMC article.
References
-
- Acuna G, Shi W, Trudeau K, Zusman DR. The ‘CheA’ and ‘CheY’ domains of Myxococcus xanthus FrzE function independently in vitro as an autokinase and a phosphate acceptor, respectively. FEBS Lett. 1995;358:31–33. - PubMed
-
- Alexander RP, Zhulin IB. Submitted for publication. 2007.
-
- Anantharaman V, Aravind L. Cache: A signaling domain common to animal Ca(2+)-channel subunits and a class of prokaryotic chemotaxis receptors. Trends Biochem. Sci. 2000;25:535–537. - PubMed
Further Reading
-
- Barnakov AN, Barnakova LA, Hazelbauer GL. Location of the receptor-interaction site on CheB, the methylesterase response regulator of bacterial chemotaxis. J. Biol. Chem. 2001;276:32984–32989. - PubMed
-
- Djordjevic S, Stock AM. Chemotaxis receptor recognition by protein methyl-transferase CheR. Nat. Struct. Biol. 1998;5:446–450. - PubMed
-
- Kall L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004;338:1027–1036. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
