

HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase
- PMID: 17630831
- PMCID: PMC1914068
- DOI: 10.1371/journal.ppat.0030085
HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase
Abstract
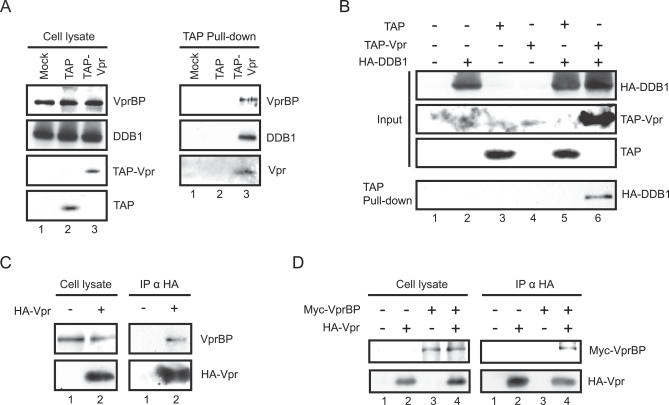
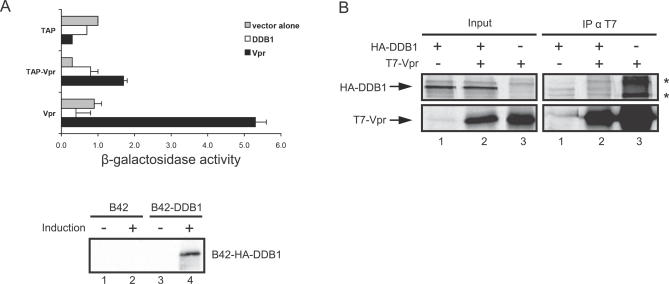
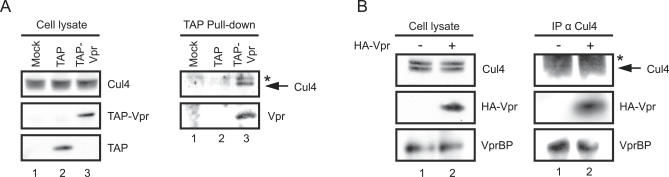
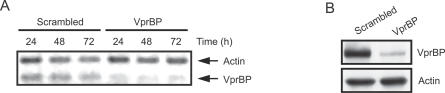
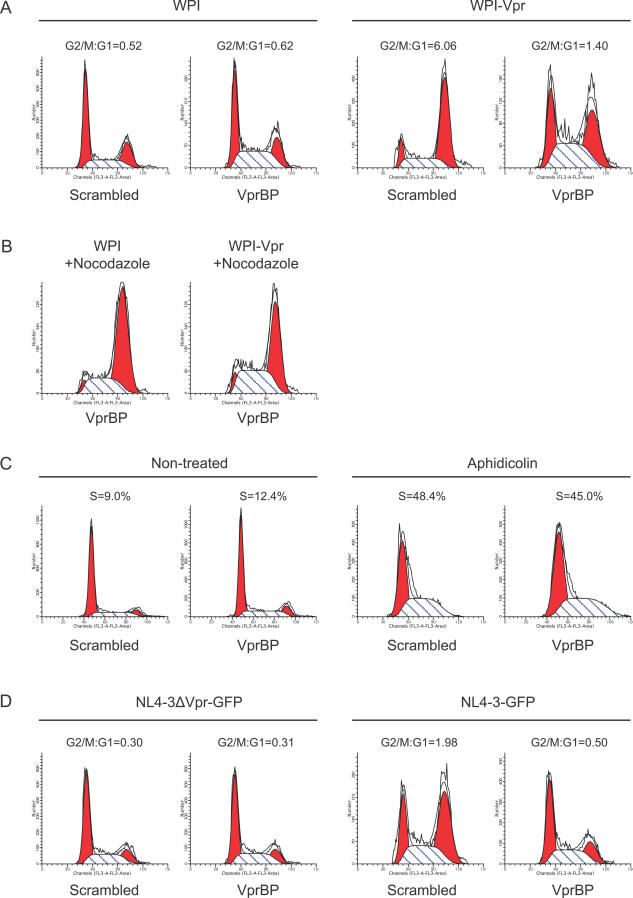
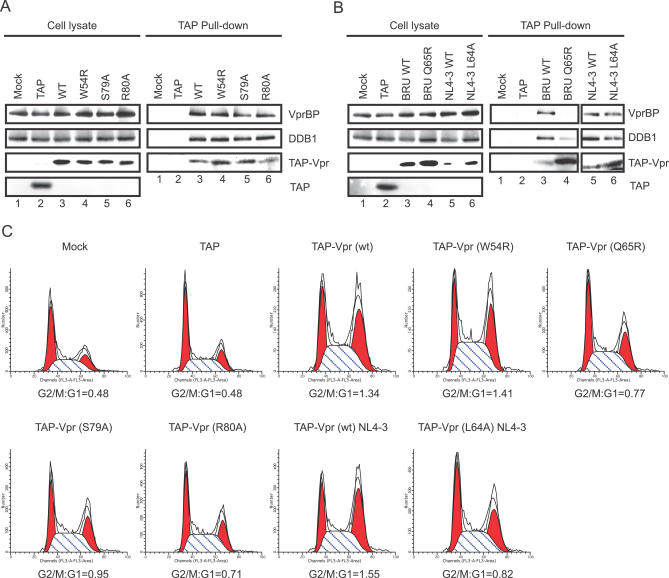
Human immunodeficiency virus type 1 (HIV-1) viral protein R (Vpr) has been shown to cause G2 cell cycle arrest in human cells by inducing ATR-mediated inactivation of p34cdc2, but factors directly engaged in this process remain unknown. We used tandem affinity purification to isolate native Vpr complexes. We found that damaged DNA binding protein 1 (DDB1), viral protein R binding protein (VPRBP), and cullin 4A (CUL4A)--components of a CUL4A E3 ubiquitin ligase complex, DDB1-CUL4A(VPRBP)--were able to associate with Vpr. Depletion of VPRBP by small interfering RNA impaired Vpr-mediated induction of G2 arrest. Importantly, VPRBP knockdown alone did not affect normal cell cycle progression or activation of ATR checkpoints, suggesting that the involvement of VPRBP in G2 arrest was specific to Vpr. Moreover, leucine/isoleucine-rich domain Vpr mutants impaired in their ability to interact with VPRBP and DDB1 also produced strongly attenuated G2 arrest. In contrast, G2 arrest-defective C-terminal Vpr mutants were found to maintain their ability to associate with these proteins, suggesting that the interaction of Vpr with the DDB1-VPRBP complex is necessary but not sufficient to block cell cycle progression. Overall, these results point toward a model in which Vpr could act as a connector between the DDB1-CUL4A(VPRBP) E3 ubiquitin ligase complex and an unknown cellular factor whose proteolysis or modulation of activity through ubiquitination would activate ATR-mediated checkpoint signaling and induce G2 arrest.
Conflict of interest statement
Figures
References
-
- Andersen JL, Planelles V. The role of Vpr in HIV-1 pathogenesis. Curr HIV Res. 2005;3:43–51. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
