

Bias in phylogenetic tree reconciliation methods: implications for vertebrate genome evolution
- PMID: 17634151
- PMCID: PMC2323230
- DOI: 10.1186/gb-2007-8-7-r141
Bias in phylogenetic tree reconciliation methods: implications for vertebrate genome evolution
Abstract
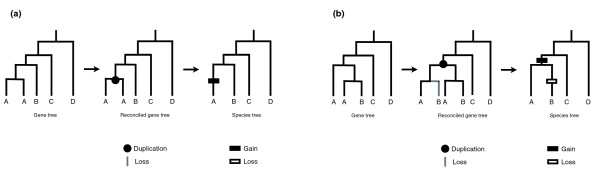
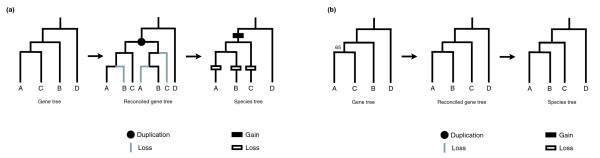
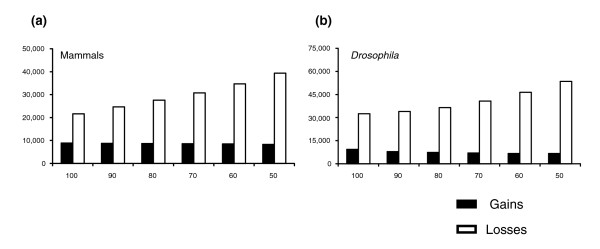
Background: Comparative genomic studies are revealing frequent gains and losses of whole genes via duplication and pseudogenization. One commonly used method for inferring the number and timing of gene gains and losses reconciles the gene tree for each gene family with the species tree of the taxa considered. Recent studies using this approach have found a large number of ancient duplications and recent losses among vertebrate genomes.
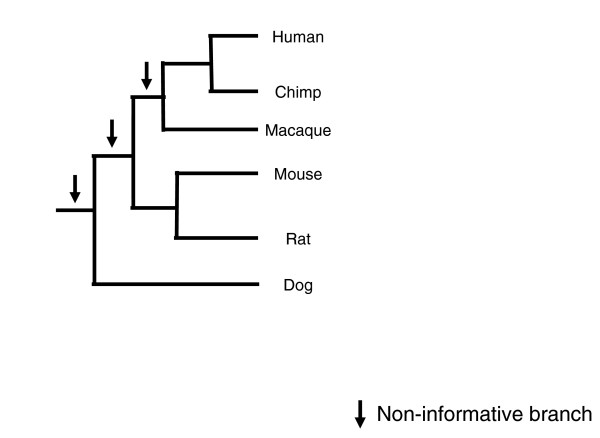
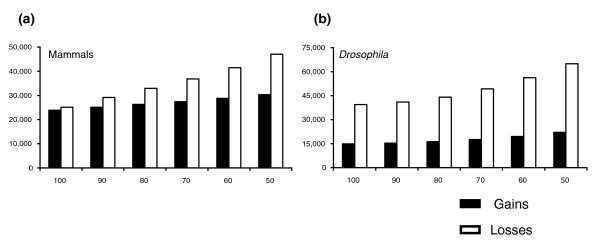
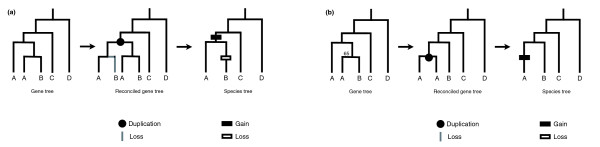
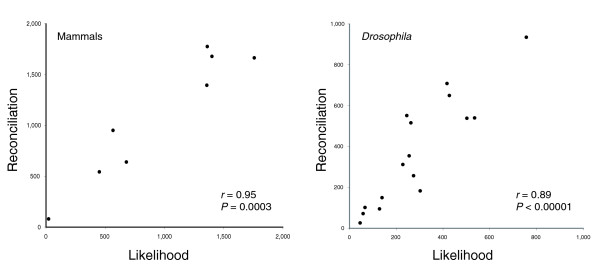
Results: I show that tree reconciliation methods are biased when the inferred gene tree is not correct. This bias places duplicates towards the root of the tree and losses towards the tips of the tree. I demonstrate that this bias is present when tree reconciliation is conducted on both multiple mammal and Drosophila genomes, and that lower bootstrap cut-off values on gene trees lead to more extreme bias. I also suggest a method for dealing with reconciliation bias, although this method only corrects for the number of gene gains on some branches of the species tree.
Conclusion: Based on the results presented, it is likely that most tree reconciliation analyses show biases, unless the gene trees used are exceptionally well-resolved and well-supported. These results cast doubt upon previous conclusions that vertebrate genome history has been marked by many ancient duplications and many recent gene losses.
Figures
References
-
- Bridges CB. The Bar "gene" a duplication. Science. 1936;83:210–211. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
