

Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons
- PMID: 17634376
- PMCID: PMC6672878
- DOI: 10.1523/JNEUROSCI.1644-07.2007
Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons
Abstract
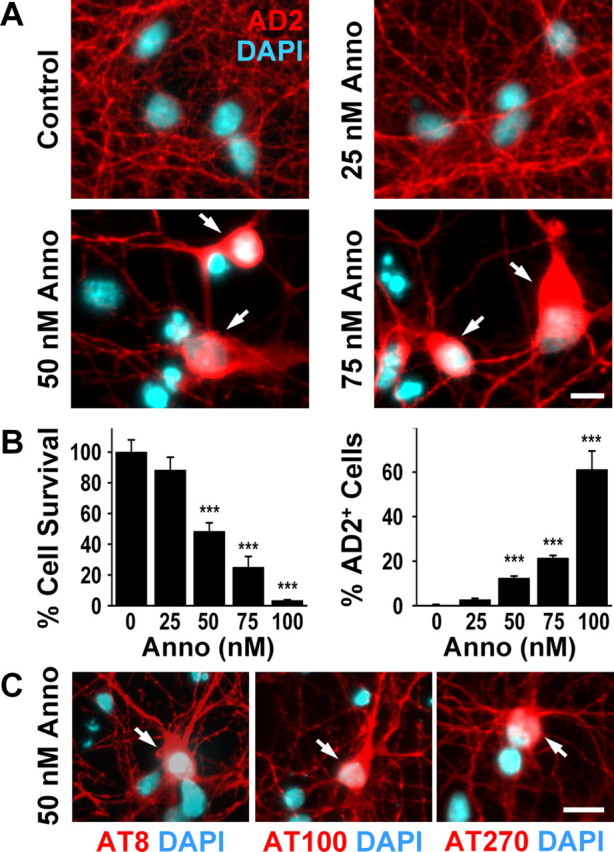
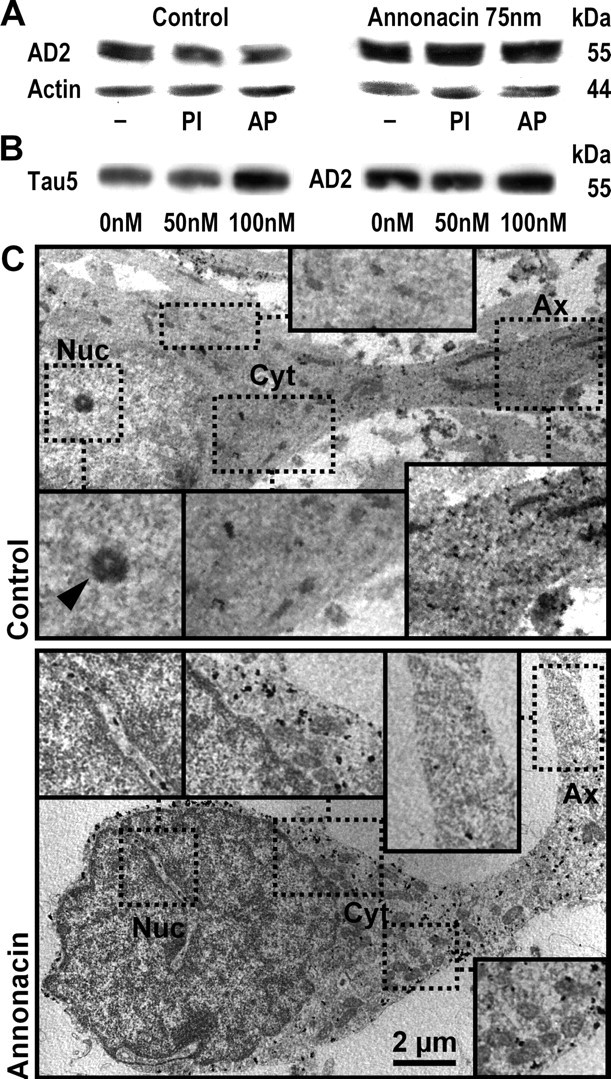
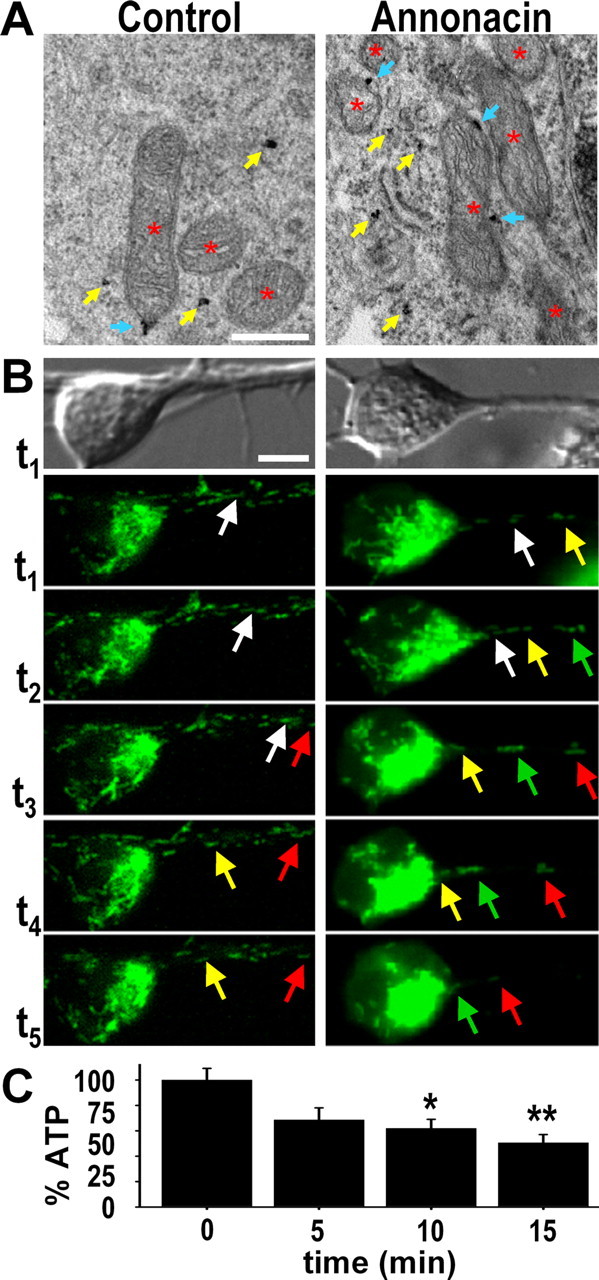
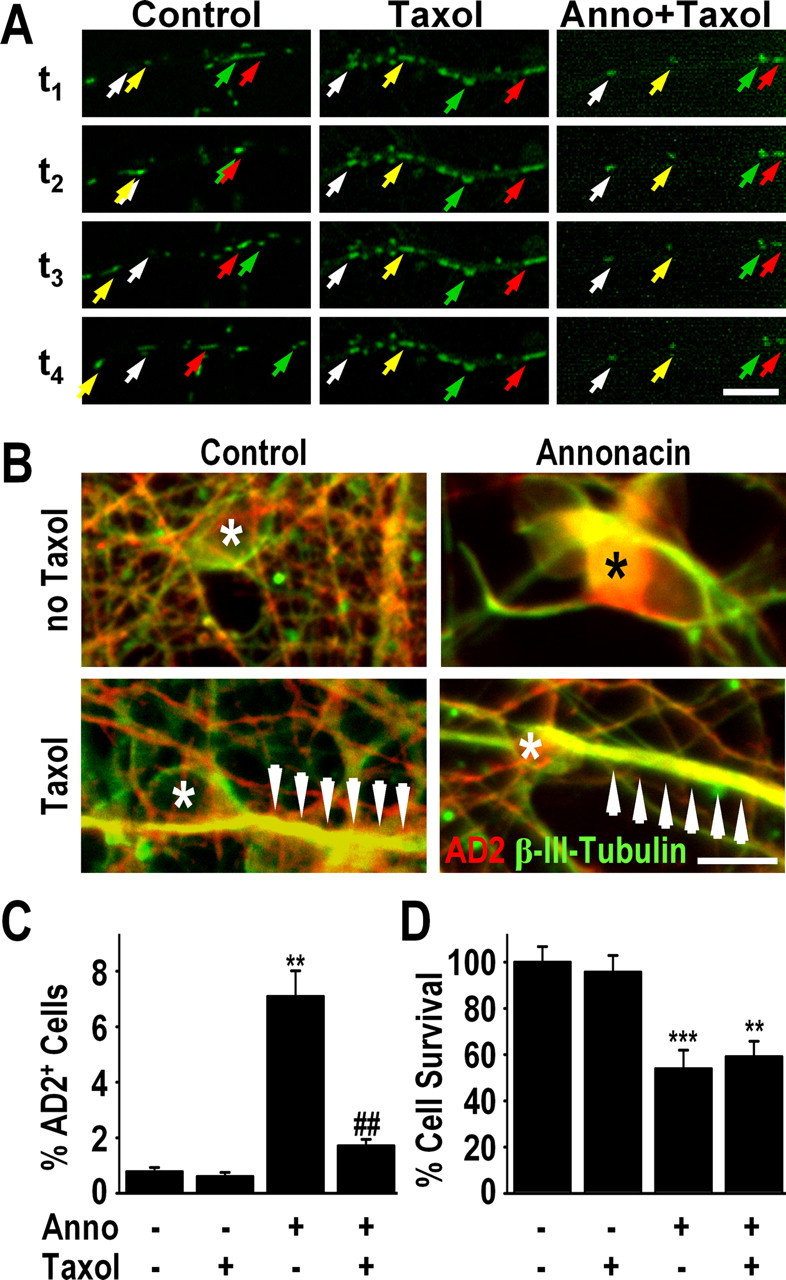
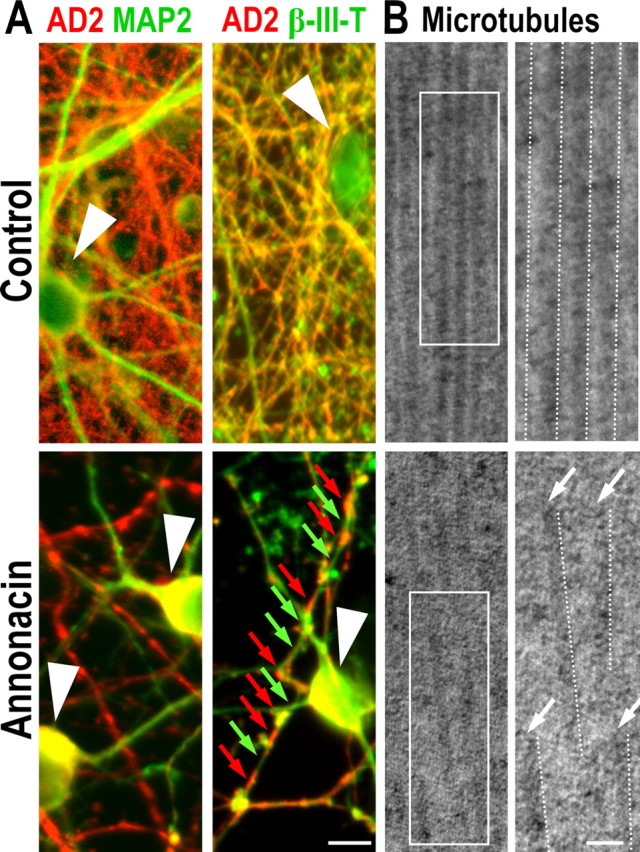
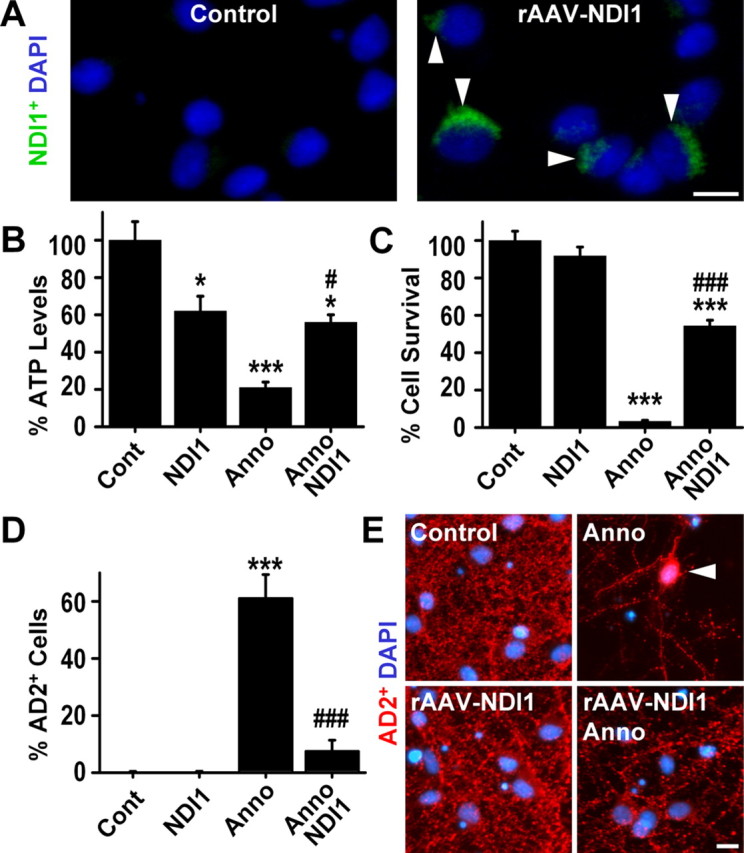
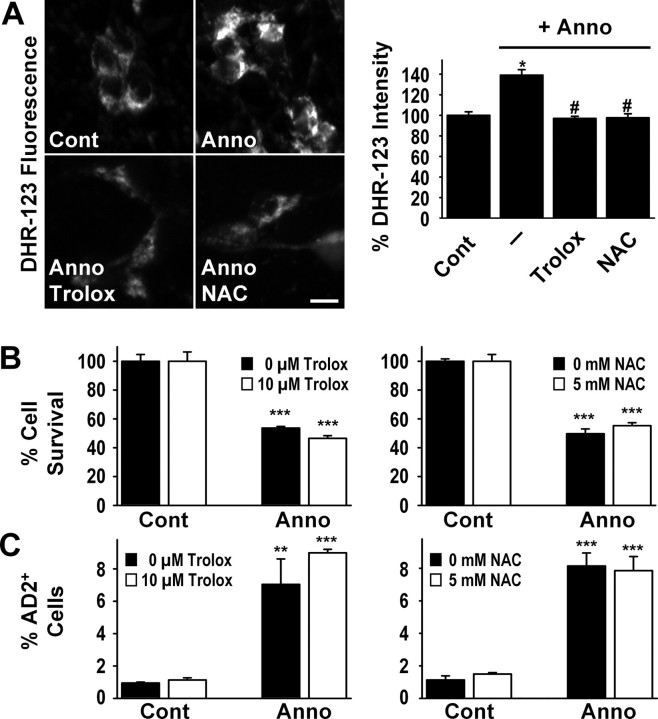
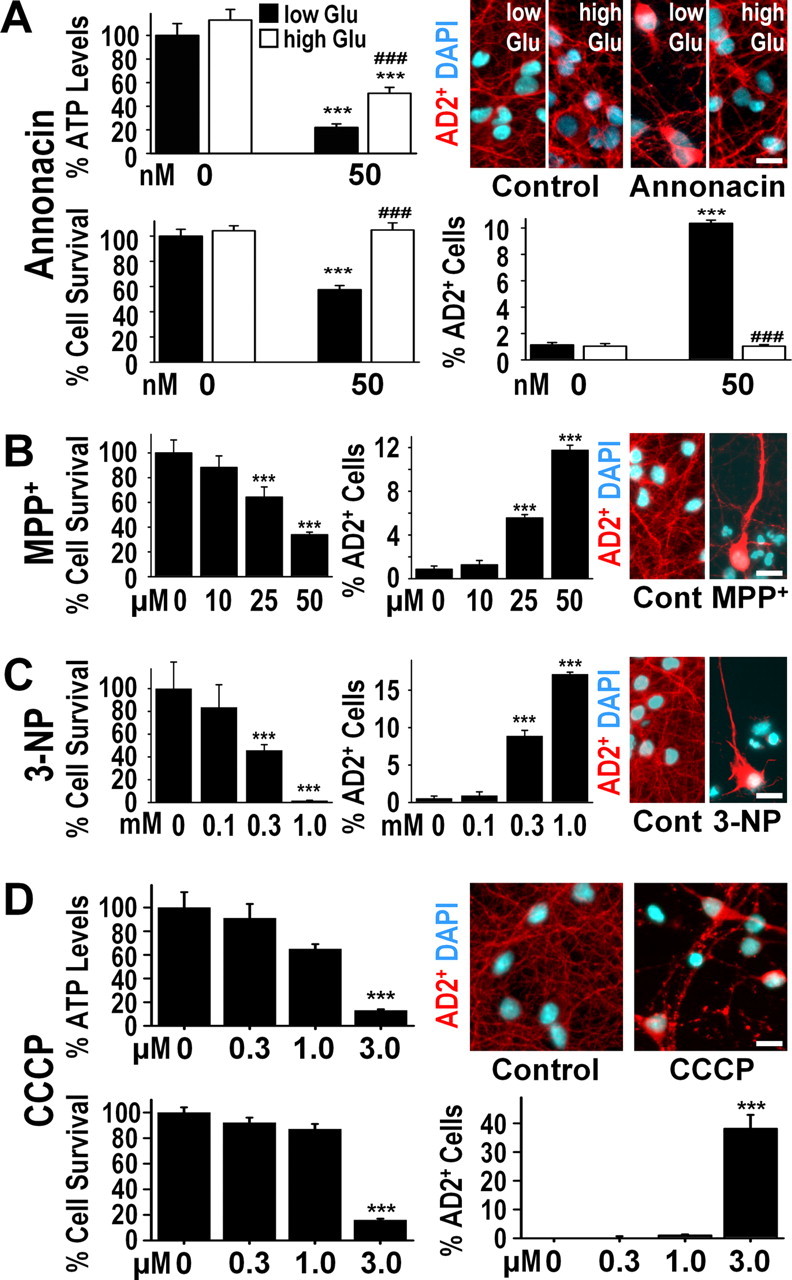
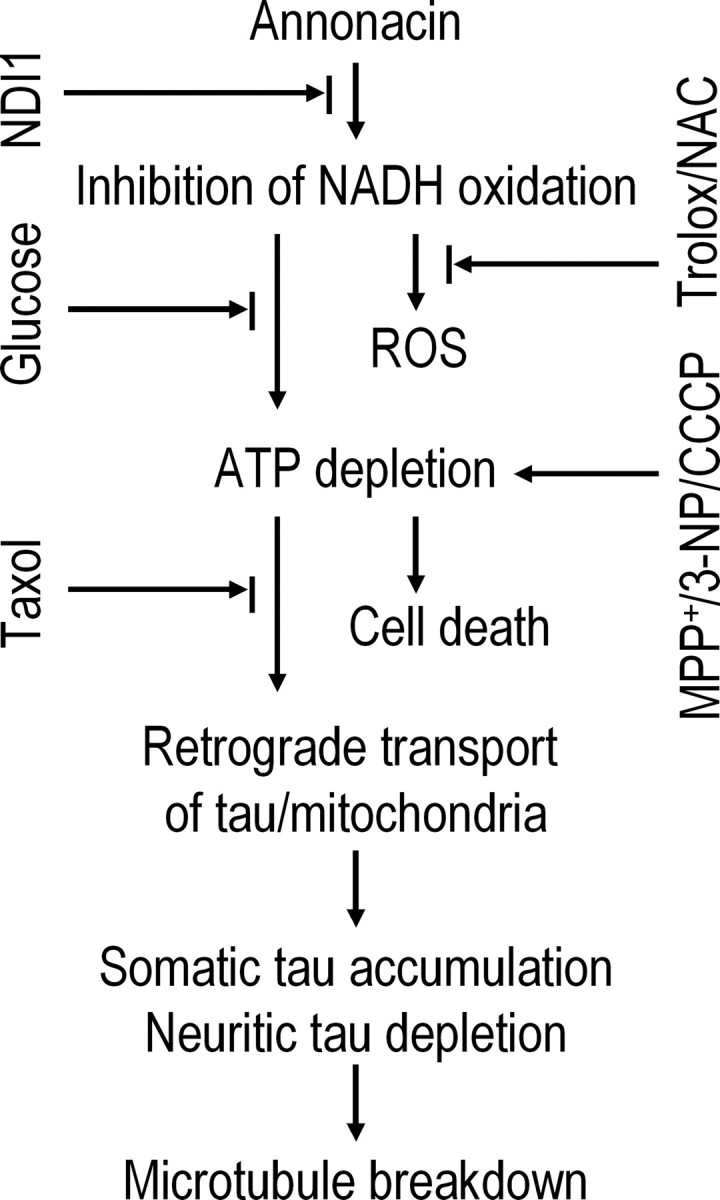
A neurodegenerative tauopathy endemic to the Caribbean island of Guadeloupe has been associated with the consumption of anonaceous plants that contain acetogenins, potent lipophilic inhibitors of complex I of the mitochondrial respiratory chain. To test the hypothesis that annonacin, a prototypical acetogenin, contributes to the etiology of the disease, we investigated whether annonacin affects the cellular distribution of the protein tau. In primary cultures of rat striatal neurons treated for 48 h with annonacin, there was a concentration-dependent decrease in ATP levels, a redistribution of tau from the axons to the cell body, and cell death. Annonacin induced the retrograde transport of mitochondria, some of which had tau attached to their outer membrane. Taxol, a drug that displaces tau from microtubules, prevented the somatic redistribution of both mitochondria and tau but not cell death. Antioxidants, which scavenged the reactive oxygen species produced by complex I inhibition, did not affect either the redistribution of tau or cell death. Both were prevented, however, by forced expression of the NDI1 nicotinamide adenine dinucleotide (NADH)-quinone-oxidoreductase of Saccharomyces cerevisiae, which can restore NADH oxidation in complex I-deficient mammalian cells and stimulation of energy production via anaerobic glycolysis. Consistently, other ATP-depleting neurotoxins (1-methyl-4-phenylpyridinium, 3-nitropropionic, and carbonyl cyanide m-chlorophenylhydrazone) reproduced the somatic redistribution of tau, whereas toxins that did not decrease ATP levels did not cause the redistribution of tau. Therefore, the annonacin-induced ATP depletion causes the retrograde transport of mitochondria to the cell soma and induces changes in the intracellular distribution of tau in a way that shares characteristics with some neurodegenerative diseases.
Figures
Similar articles
-
The mitochondrial complex I inhibitor annonacin is toxic to mesencephalic dopaminergic neurons by impairment of energy metabolism.Neuroscience. 2003;121(2):287-96. doi: 10.1016/s0306-4522(03)00441-x. Neuroscience. 2003. PMID: 14521988
-
Annonacin, a natural lipophilic mitochondrial complex I inhibitor, increases phosphorylation of tau in the brain of FTDP-17 transgenic mice.Exp Neurol. 2014 Mar;253:113-25. doi: 10.1016/j.expneurol.2013.12.017. Epub 2014 Jan 3. Exp Neurol. 2014. PMID: 24389273
-
Natural lipophilic inhibitors of mitochondrial complex I are candidate toxins for sporadic neurodegenerative tau pathologies.Exp Neurol. 2009 Nov;220(1):133-42. doi: 10.1016/j.expneurol.2009.08.004. Epub 2009 Aug 13. Exp Neurol. 2009. PMID: 19682988
-
Complex I: inhibitors, inhibition and neurodegeneration.Exp Neurol. 2010 Aug;224(2):331-5. doi: 10.1016/j.expneurol.2010.03.028. Epub 2010 Apr 1. Exp Neurol. 2010. PMID: 20362572 Review.
-
Atypical parkinsonism in the Caribbean island of Guadeloupe: etiological role of the mitochondrial complex I inhibitor annonacin.Mov Disord. 2008 Nov 15;23(15):2122-8. doi: 10.1002/mds.22300. Mov Disord. 2008. PMID: 18816693 Review.
Cited by
-
Disruption of mitochondrial DNA replication in Drosophila increases mitochondrial fast axonal transport in vivo.PLoS One. 2009 Nov 17;4(11):e7874. doi: 10.1371/journal.pone.0007874. PLoS One. 2009. PMID: 19924234 Free PMC article.
-
Rapid changes in phospho-MAP/tau epitopes during neuronal stress: cofilin-actin rods primarily recruit microtubule binding domain epitopes.PLoS One. 2011;6(6):e20878. doi: 10.1371/journal.pone.0020878. Epub 2011 Jun 28. PLoS One. 2011. PMID: 21738590 Free PMC article.
-
Analysis of cytotoxic activity at short incubation times reveals profound differences among Annonaceus acetogenins, inhibitors of mitochondrial Complex I.J Bioenerg Biomembr. 2013 Feb;45(1-2):145-52. doi: 10.1007/s10863-012-9490-8. Epub 2012 Nov 21. J Bioenerg Biomembr. 2013. PMID: 23180141
-
Mechanisms of Neurodegeneration in Various Forms of Parkinsonism-Similarities and Differences.Cells. 2021 Mar 16;10(3):656. doi: 10.3390/cells10030656. Cells. 2021. PMID: 33809527 Free PMC article. Review.
-
Evaluation of Genotoxicity and Toxicity of Annona muricata L. Seeds and In Silico Studies.Molecules. 2022 Dec 27;28(1):231. doi: 10.3390/molecules28010231. Molecules. 2022. PMID: 36615425 Free PMC article.
References
-
- Albers DS, Swerdlow RH, Manfredi G, Gajewski C, Yang L, Parker WD, Jr, Beal MF. Further evidence for mitochondrial dysfunction in progressive supranuclear palsy. Exp Neurol. 2001;168:196–198. - PubMed
-
- Angibaud G, Gaultier C, Rascol O. Atypical parkinsonism and Annonaceae consumption in New Caledonia. Mov Disord. 2004;19:603–604. - PubMed
-
- Arriagada PV, Growdon JH, Hedley-White ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;82:239–259. - PubMed
-
- Bai Y, Hajek P, Chomyn A, Chan E, Seo BB, Matsuno-Yagi A, Yagi T, Attardi G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J Biol Chem. 2001;276:38808–38813. - PubMed
-
- Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989;477:90–99. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases