

Distinct mechanisms of cardiomyocyte apoptosis induced by doxorubicin and hypoxia converge on mitochondria and are inhibited by Bcl-xL
- PMID: 17635642
- PMCID: PMC3922357
- DOI: 10.1111/j.1582-4934.2007.00042.x
Distinct mechanisms of cardiomyocyte apoptosis induced by doxorubicin and hypoxia converge on mitochondria and are inhibited by Bcl-xL
Abstract
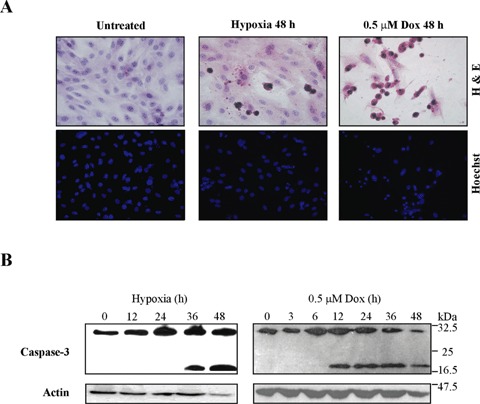
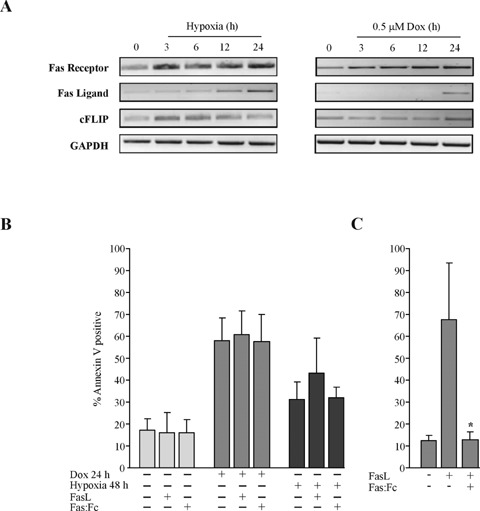
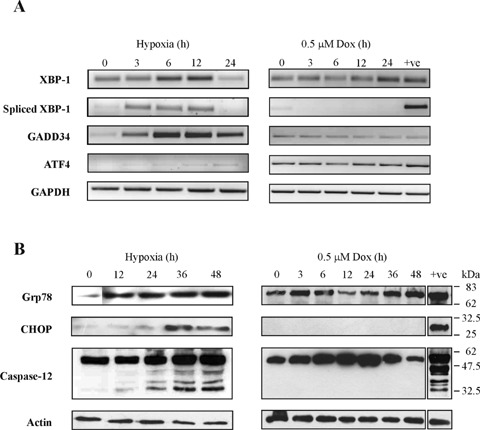
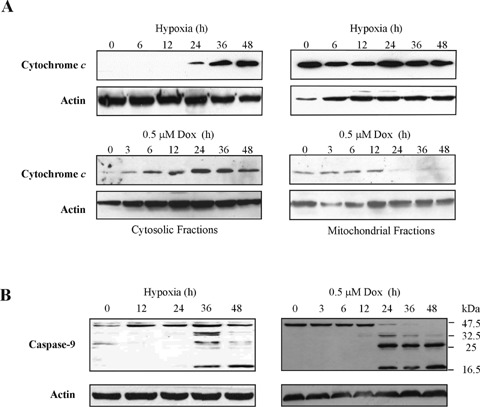
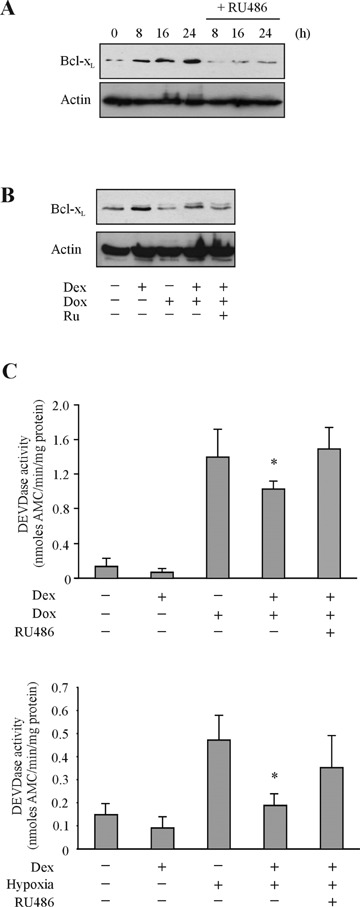
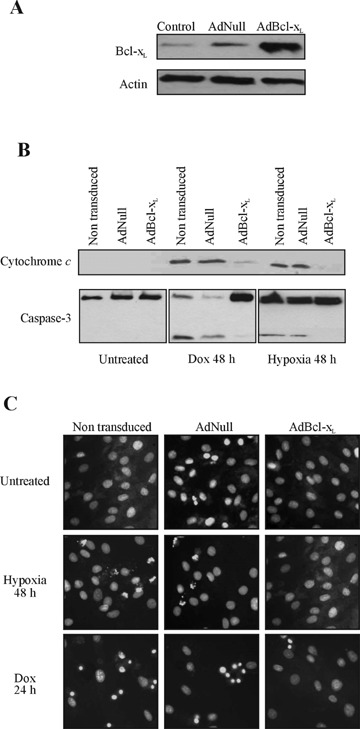
Hypoxia and doxorubicin can cause cardiotoxicity and loss of myocardial function. These effects are due, in part, to an induction of apoptosis. Herein we identify the apoptotic pathways activated in H9c2 cells in response to hypoxia (O(2)/N(2)/CO(2), 0.5:94.5:5) and doxorubicin (0.5 muM). Although the apoptosis induced was accompanied by induction of Fas and Fas ligand, the death receptor pathway was not critical for caspase activation by either stimulus. Hypoxia induced the expression of endoplasmic reticulum (ER) stress mediators and processed ER-resident pro-caspase-12 whereas doxorubicin did not induce an ER stress response. Most importantly, both stimuli converged on mitochondria to promote apoptosis. Accumulation of cytochrome c in the cytosol coincided with the processing of pro-caspase-9 and -3. Increasing the expression of the anti-apoptotic protein Bcl-x(L), either by dexamethasone or adenovirus-mediated transduction, protected H9c2 cells from doxorubicin- and hypoxia-induced apoptosis. Bcl-x(L) attenuated mitochondrial cytochrome crelease and reduced downstream pro-caspase processing and apoptosis. These data demonstrate that two distinct cardiomyocyte-damaging stimuli converge on mitochondria thus presenting this organelle as a potentially important therapeutic target for anti-apoptotic strategies for cardiovascular diseases.
Figures
Similar articles
-
C-reactive protein augments hypoxia-induced apoptosis through mitochondrion-dependent pathway in cardiac myocytes.Mol Cell Biochem. 2008 Mar;310(1-2):215-26. doi: 10.1007/s11010-007-9683-3. Epub 2007 Dec 29. Mol Cell Biochem. 2008. PMID: 18165866
-
Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria- and Fas/FasL-mediated pathways.Toxicol In Vitro. 2011 Aug;25(5):1027-32. doi: 10.1016/j.tiv.2011.03.023. Epub 2011 Apr 3. Toxicol In Vitro. 2011. PMID: 21466843
-
ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity.J Mol Med (Berl). 2009 Apr;87(4):401-10. doi: 10.1007/s00109-008-0434-z. Epub 2009 Jan 13. J Mol Med (Berl). 2009. PMID: 19139834
-
TanshinoneIIA and cryptotanshinone protect against hypoxia-induced mitochondrial apoptosis in H9c2 cells.PLoS One. 2013;8(1):e51720. doi: 10.1371/journal.pone.0051720. Epub 2013 Jan 14. PLoS One. 2013. PMID: 23341883 Free PMC article.
-
MicroRNA-29b Regulates the Mitochondria-Dependent Apoptotic Pathway by Targeting Bax in Doxorubicin Cardiotoxicity.Cell Physiol Biochem. 2018;48(2):692-704. doi: 10.1159/000491896. Epub 2018 Jul 19. Cell Physiol Biochem. 2018. PMID: 30025410
Cited by
-
Critical role of nuclear calcium/calmodulin-dependent protein kinase IIdeltaB in cardiomyocyte survival in cardiomyopathy.J Biol Chem. 2009 Sep 11;284(37):24857-68. doi: 10.1074/jbc.M109.003186. Epub 2009 Jul 14. J Biol Chem. 2009. PMID: 19602725 Free PMC article.
-
Oral treatment with the herbal formula B307 alleviates cardiac toxicity in doxorubicin-treated mice via suppressing oxidative stress, inflammation, and apoptosis.Onco Targets Ther. 2015 May 26;8:1193-210. doi: 10.2147/OTT.S82936. eCollection 2015. Onco Targets Ther. 2015. PMID: 26060405 Free PMC article.
-
Acidic pre-conditioning suppresses apoptosis and increases expression of Bcl-xL in coronary endothelial cells under simulated ischaemia.J Cell Mol Med. 2008 Sep-Oct;12(5A):1584-92. doi: 10.1111/j.1582-4934.2007.00172.x. Epub 2007 Nov 29. J Cell Mol Med. 2008. PMID: 18053090 Free PMC article.
-
Complex patterns of mitochondrial dynamics in human pancreatic cells revealed by fluorescent confocal imaging.J Cell Mol Med. 2010 Jan;14(1-2):417-25. doi: 10.1111/j.1582-4934.2009.00750.x. Epub 2009 Mar 27. J Cell Mol Med. 2010. PMID: 19382913 Free PMC article.
-
Cardioprotective effects of sitagliptin against doxorubicin-induced cardiotoxicity in rats.Exp Biol Med (Maywood). 2016 Aug;241(14):1577-87. doi: 10.1177/1535370216643418. Epub 2016 Apr 1. Exp Biol Med (Maywood). 2016. PMID: 27037281 Free PMC article.
References
-
- Gill C, Mestril R, Samali A. Losing heart: the role of apoptosis in heart disease–a novel therapeutic target? FASEB J. 2002;16:135–46. - PubMed
-
- Logue SE, Gustafsson AB, Samali A, Gottlieb RA. Ischaemia/reperfusion injury at the intersection with cell death. J Mol Cell Cardiol. 2005;38:21–33. - PubMed
-
- Samali A, Zhivotovsky B, Jones D, Nagata S, Orrenius S. Apoptosis: cell death defined by caspase activation. Cell Death Differ. 1999;6:495–6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
