

Automated recognition of retroviral sequences in genomic data--RetroTector
- PMID: 17636050
- PMCID: PMC1976444
- DOI: 10.1093/nar/gkm515
Automated recognition of retroviral sequences in genomic data--RetroTector
Abstract
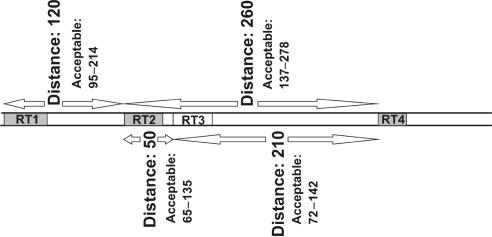
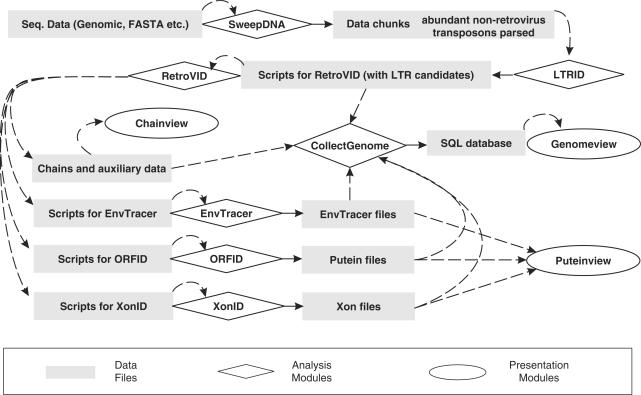
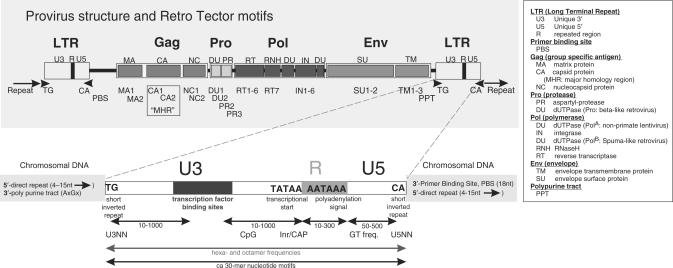
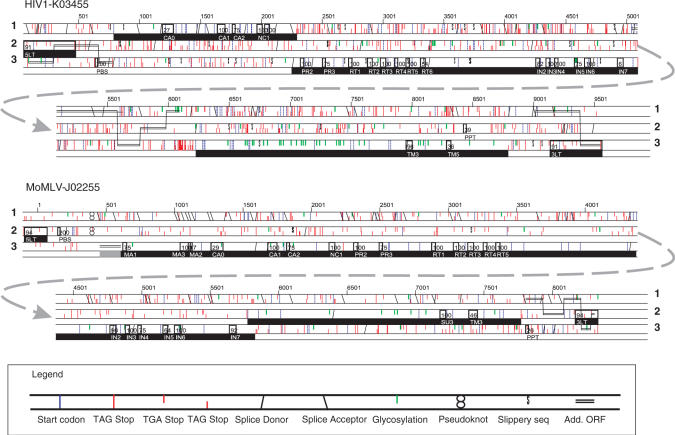
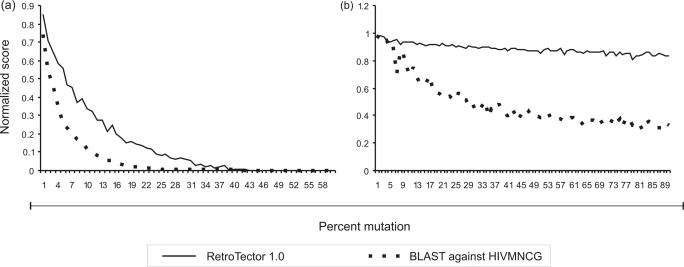
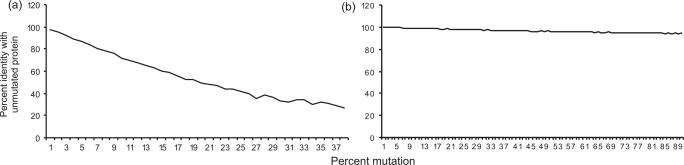
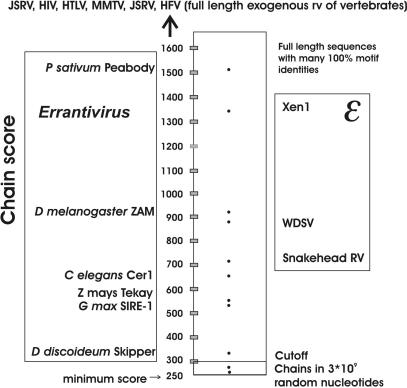
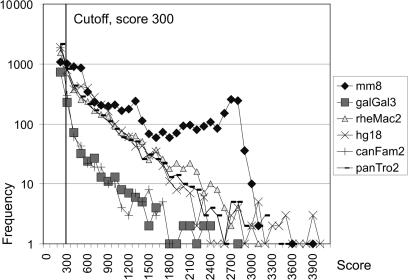
Eukaryotic genomes contain many endogenous retroviral sequences (ERVs). ERVs are often severely mutated, therefore difficult to detect. A platform independent (Java) program package, RetroTector (ReTe), was constructed. It has three basic modules: (i) detection of candidate long terminal repeats (LTRs), (ii) detection of chains of conserved retroviral motifs fulfilling distance constraints and (iii) attempted reconstruction of original retroviral protein sequences, combining alignment, codon statistics and properties of protein ends. Other features are prediction of additional open reading frames, automated database collection, graphical presentation and automatic classification. ReTe favors elements >1000-bp long due to its dependence on order of and distances between retroviral fragments. It detects single or low-copy-number elements. ReTe assigned a 'retroviral' score of 890-2827 to 10 exogenous retroviruses from seven genera, and accurately predicted their genes. In a simulated model, ReTe was robust against mutational decay. The human genome was analyzed in 1-2 days on a LINUX cluster. Retroviral sequences were detected in divergent vertebrate genomes. Most ReTe detected chains were coincident with Repeatmasker output and the HERVd database. ReTe did not report most of the evolutionary old HERV-L related and MalR sequences, and is not yet tailored for single LTR detection. Nevertheless, ReTe rationally detects and annotates many retroviral sequences.
Figures
References
-
- Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. New York, USA: Cold Spring Harbor Laboratory Press; 1997. - PubMed
-
- IHGSC. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- CSAC. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. - PubMed
-
- ICGSC. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. - PubMed
-
- Kumar A, Bennetzen JL. Plant retrotransposons. Annu. Rev. Genet. 1999;33:479–532. - PubMed
