

Anti-inflammatory effects of phosphatidylcholine
- PMID: 17636253
- PMCID: PMC2693065
- DOI: 10.1074/jbc.M704408200
Anti-inflammatory effects of phosphatidylcholine
Abstract
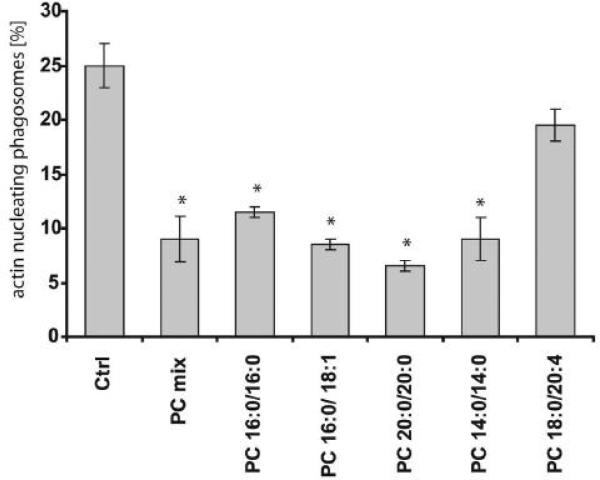
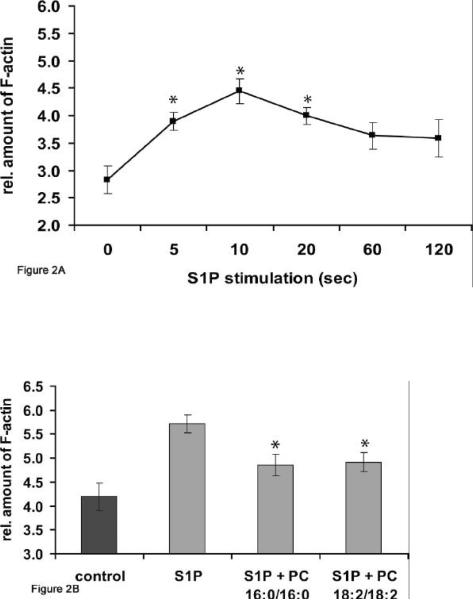
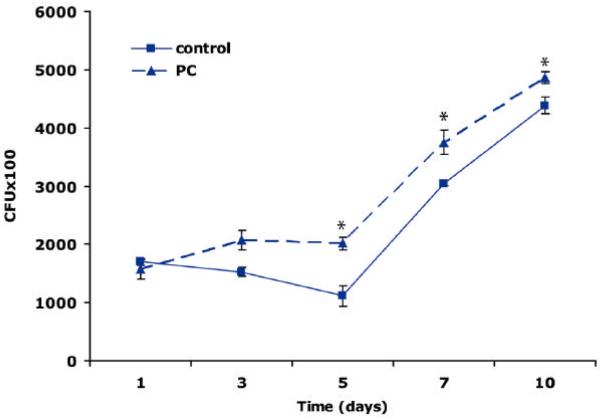
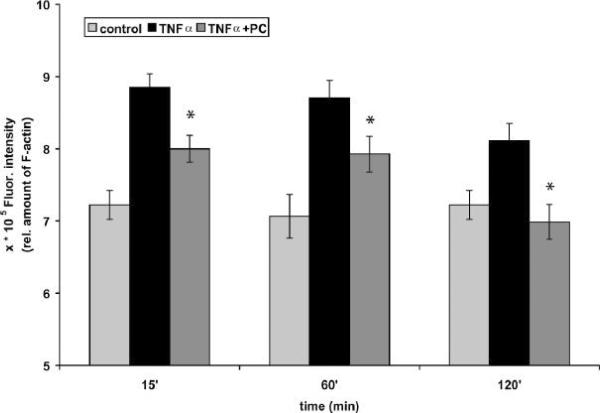
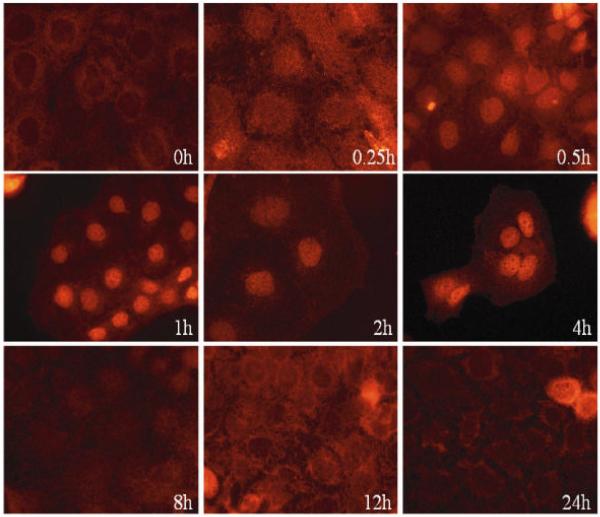
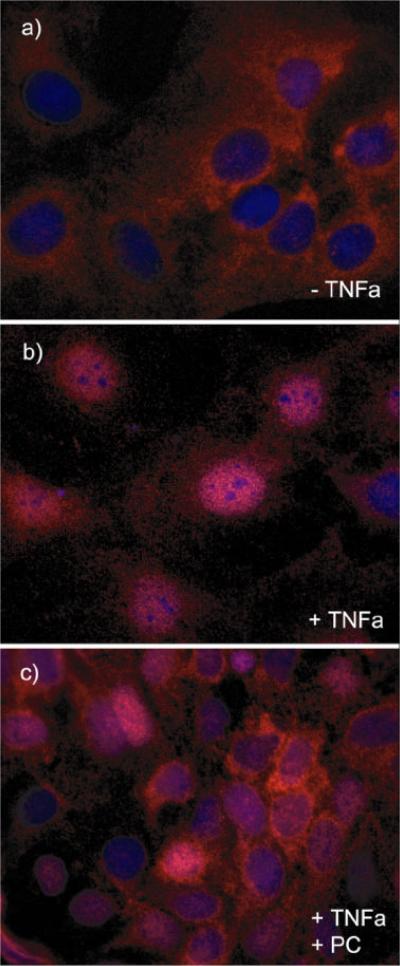
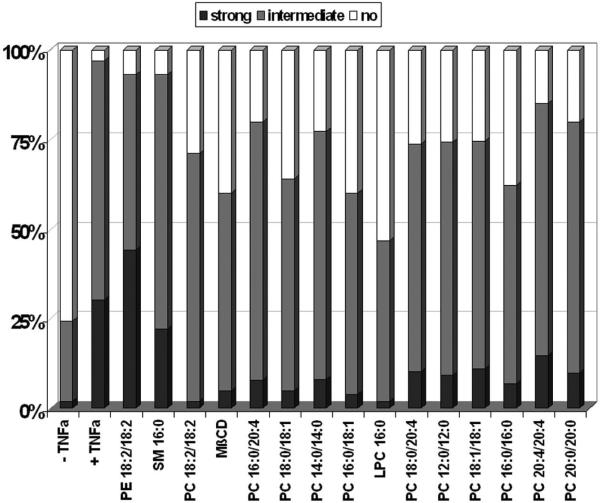
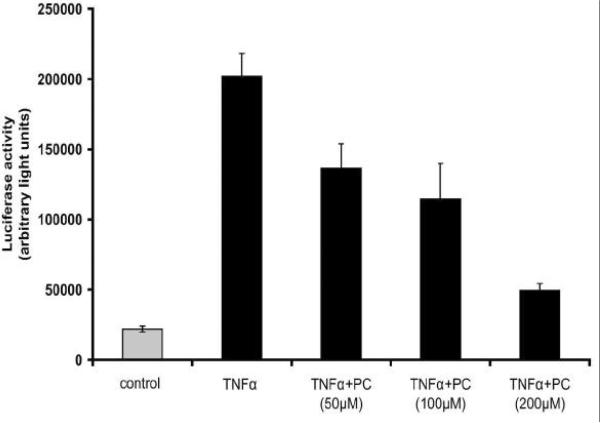
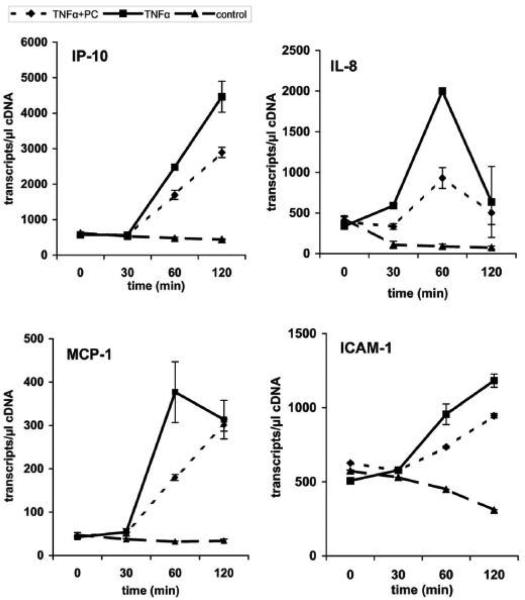
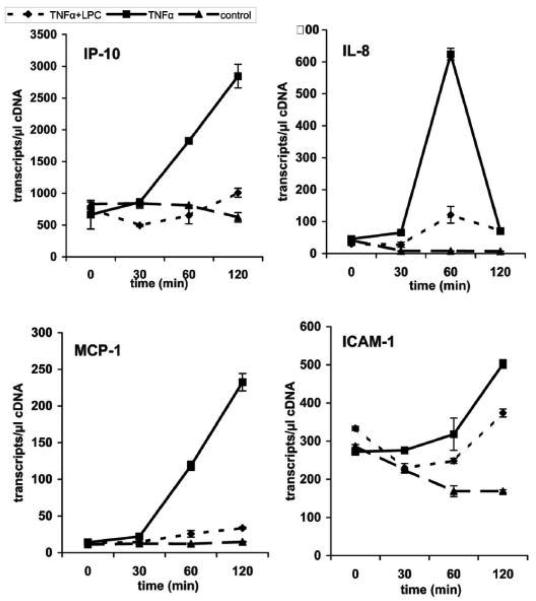
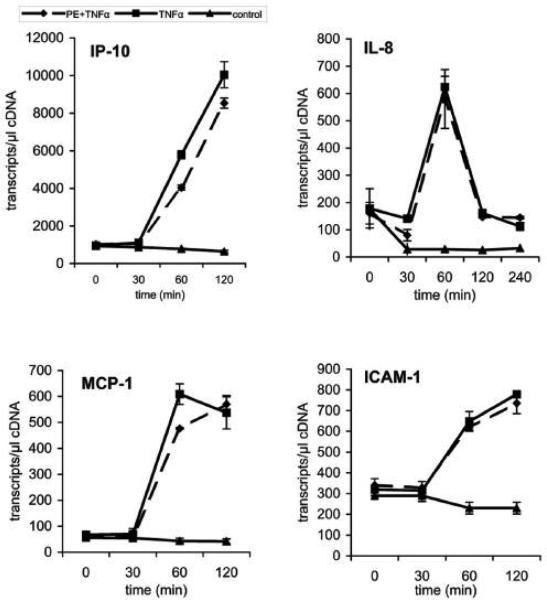
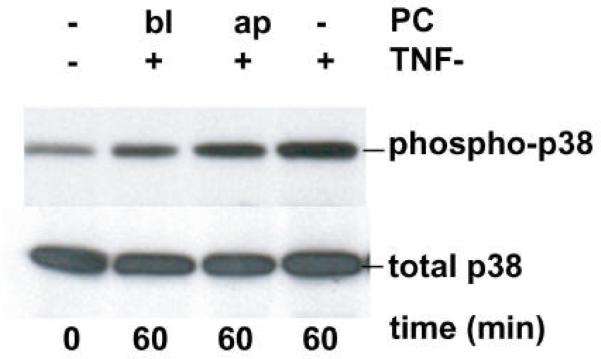
We recently showed that mucus from patients with ulcerative colitis, a chronic inflammatory disorder of the colon, is characterized by a low level of phosphatidylcholine (PC) while clinical studies reveal that therapeutic addition of PC using slow release preparations is beneficial. The positive role of PC in this disease is still elusive. Here we tested the hypothesis that exogenous application of PC has anti-inflammatory properties using three model systems. First, human Caco-2 cells were treated with tumor necrosis factor-alpha (TNF-alpha) to induce a pro-inflammatory response via activation of NF-kappaB. Second, latex bead phagosomes were analyzed for their ability to assemble actin in vitro, a process linked to pro-inflammatory signaling and correlating with the growth versus killing of mycobacteria in macrophages. The third system used was the rapid assembly of plasma membrane actin in macrophages in response to sphingosine 1-phosphate. TNF-alpha induced a pro-inflammatory response in Caco-2 cells, including 1) assembly of plasma membrane actin; 2) activation of both MAPKs ERK and p38; 3) transport of NF-kappaB subunits to the nucleus; and 4) subsequent up-regulation of the synthesis of pro-inflammatory gene products. Exogenous addition of most PCs tested significantly inhibited these processes. Other phospholipids like sphingomyelin or phosphatidylethanolamine showed no effects in these assays. PC also inhibited latex bead phagosome actin assembly, the killing of Mycobacterium tuberculosis in macrophages, and the sphingosine 1-phosphate-induced actin assembly in macrophages. TNF-alpha induces the activation of signaling molecules and the reorganization of the actin cytoskeleton in human intestinal cells. Exogenous application of PC blocks pro-inflammatory signaling in Caco-2 cells, in phagosomes in vitro and facilitates intracellular survival of mycobacteria. We provide further evidence that actin assembly by membranes is part of the pro-inflammatory response. Collectively, these results provide a molecular foundation for the clinical studies showing a beneficial effect of PC therapy in ulcerative colitis.
Figures

References
-
- Maaser C, Kagnoff MF. Z Gastroenterol. 2002;40:525–529. - PubMed
-
- Wallace JL, Granger DN. Faseb J. 1996;10:731–740. - PubMed
-
- Lichtenberger LM. Annu Rev Physiol. 1995;57:565–583. - PubMed
-
- Kao YC, Lichtenberger LM. Gastroenterology. 1991;101:7–21. - PubMed
-
- Bengmark S, Jeppsson B. JPEN J Parenter Enteral Nutr. 1995;19:410–415. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
