

The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside
- PMID: 17636426
- PMCID: PMC2847875
- DOI: 10.1007/s10689-007-9145-9
The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside
Abstract
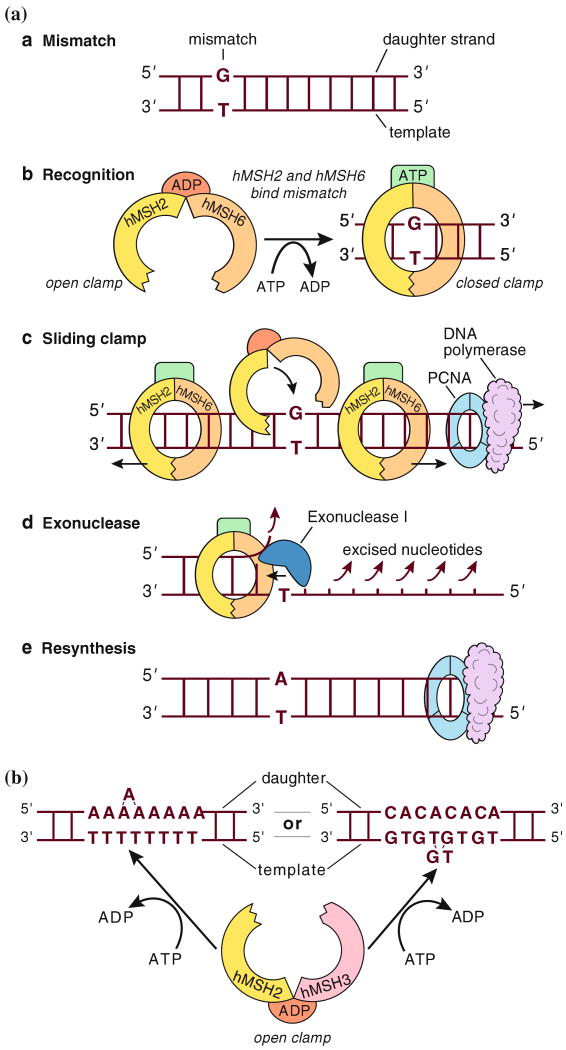
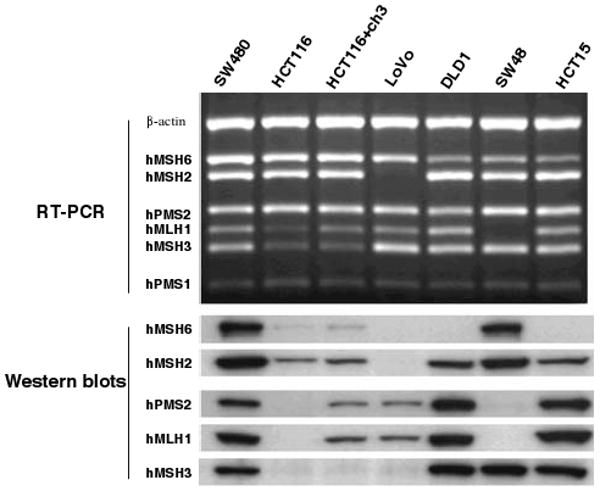
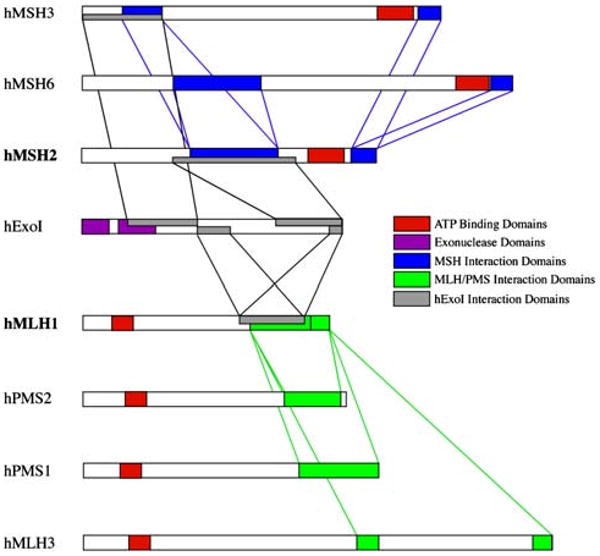
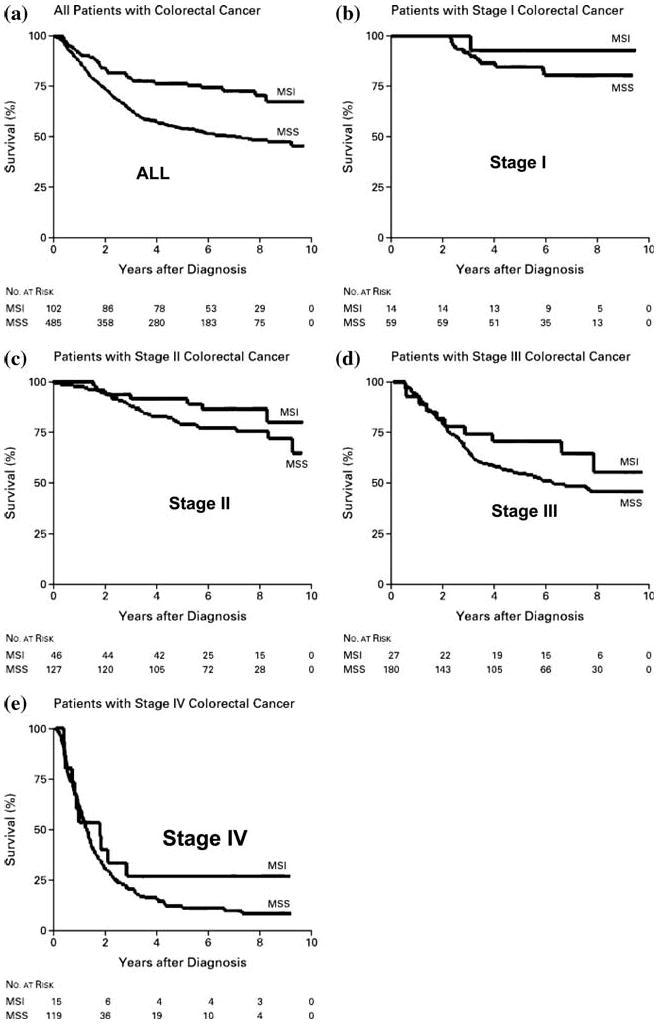
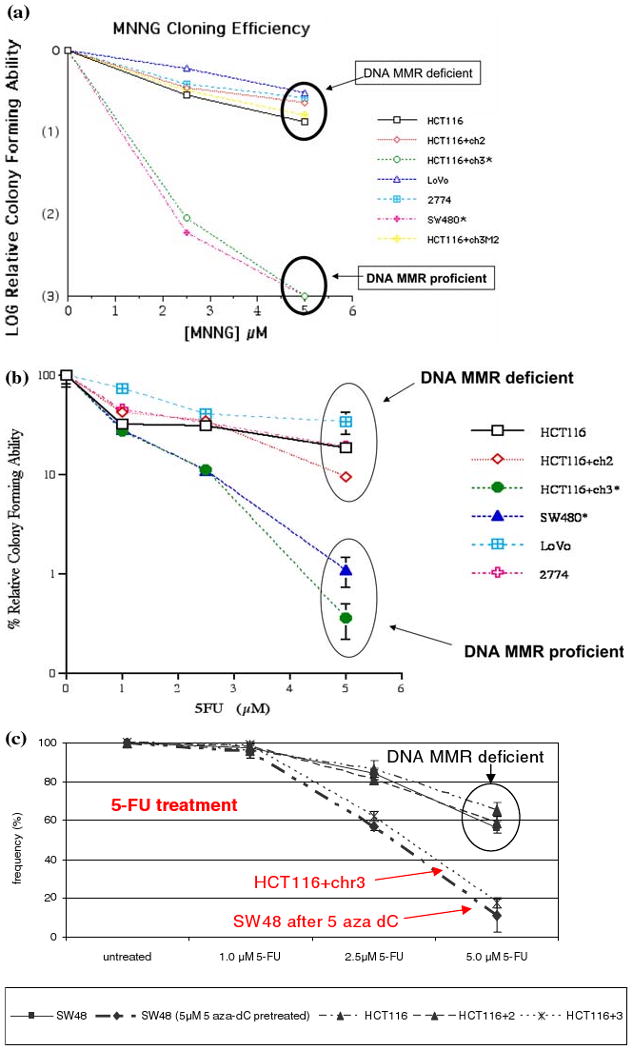
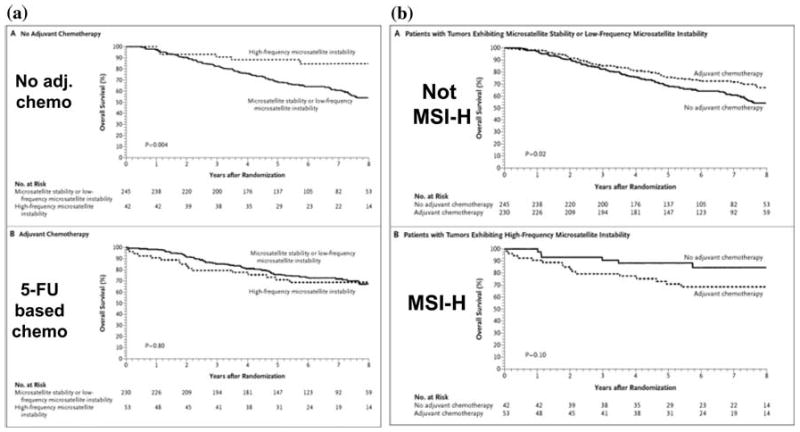
Lynch syndrome is an inherited disease caused by a germline mutation in one of four DNA mismatch repair (MMR) genes. The clinical manifestations can be somewhat variable depending upon which gene is involved, and where the mutation occurs. Moreover, the approach to the diagnosis of Lynch syndrome is becoming more complex as more is learned about the disease, and one needs to understand how the DNA MMR proteins function, and what makes them malfunction, to have an optimal appreciation of how to interpret diagnostic studies such as microsatellite instability and immunohistochemistry of the DNA MMR proteins. Finally, an understanding of the role of the DNA MMR system in regulation of the cell cycle and the response to DNA damage helps illuminate the differences in natural history and response to chemotherapeutic agents seen in Lynch syndrome.
Figures

References
-
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–932. - PubMed
-
- Gradia S, Subramanian D, Wilson T, et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3(2):255–261. - PubMed
-
- Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126(2):297–308. - PubMed
-
- Heinen CD, Schmutte C, Fishel R. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol Ther. 2002;1(5):477–485. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
