

Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome
- PMID: 17638852
- PMCID: PMC2018675
- DOI: 10.1182/blood-2006-12-061507
Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome
Erratum in
- Blood. 2008 Jan 1;111(1):49
Abstract
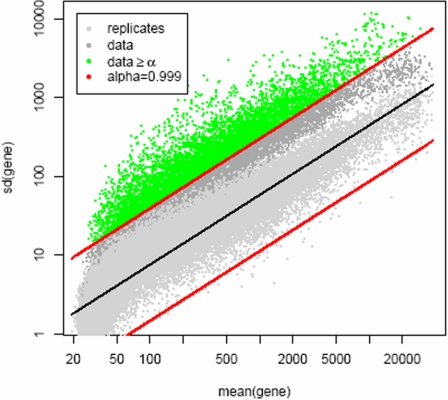
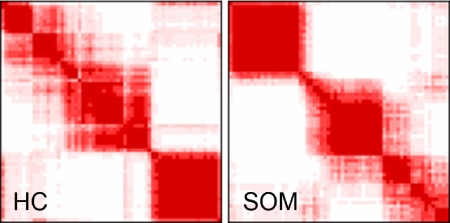
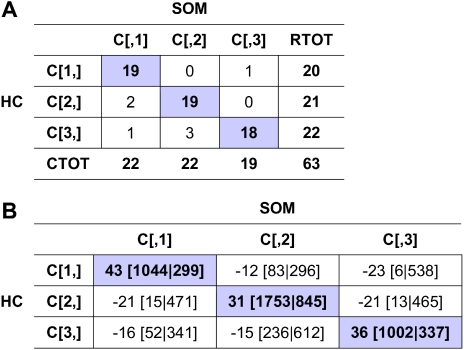
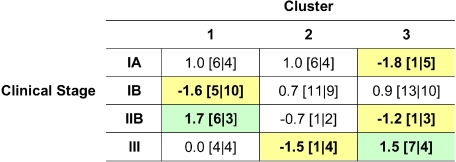
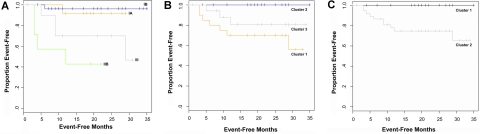
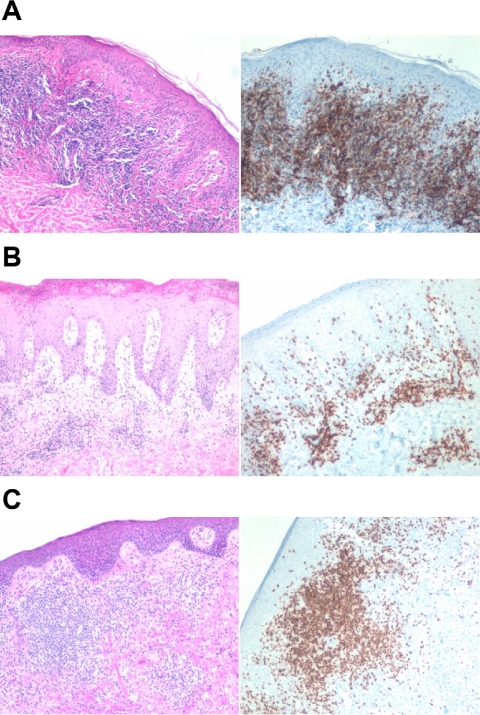
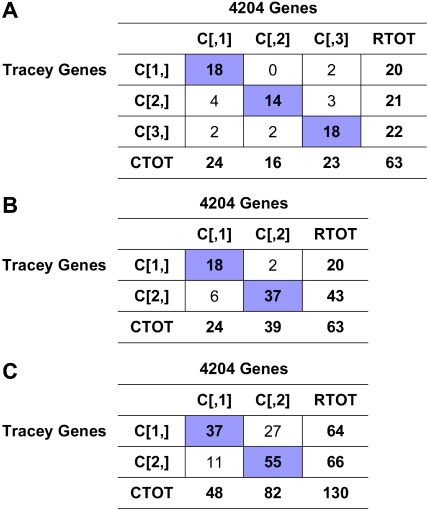
Cutaneous T-cell lymphoma (CTCL) is defined by infiltration of activated and malignant T cells in the skin. The clinical manifestations and prognosis in CTCL are highly variable. In this study, we hypothesized that gene expression analysis in lesional skin biopsies can improve understanding of the disease and its management. Based on 63 skin samples, we performed consensus clustering, revealing 3 patient clusters. Of these, 2 clusters tended to differentiate limited CTCL (stages IA and IB) from more extensive CTCL (stages IB and III). Stage IB patients appeared in both clusters, but those in the limited CTCL cluster were more responsive to treatment than those in the more extensive CTCL cluster. The third cluster was enriched in lymphocyte activation genes and was associated with a high proportion of tumor (stage IIB) lesions. Survival analysis revealed significant differences in event-free survival between clusters, with poorest survival seen in the activated lymphocyte cluster. Using supervised analysis, we further characterized genes significantly associated with lower-stage/treatment-responsive CTCL versus higher-stage/treatment-resistant CTCL. We conclude that transcriptional profiling of CTCL skin lesions reveals clinically relevant signatures, correlating with differences in survival and response to treatment. Additional prospective long-term studies to validate and refine these findings appear warranted.
Figures


References
-
- Shin J, Jones DA, Hurwitz D, Kupper TS. Gene expression profiling in cutaneous T cell lymphoma skin lesions. J Invest Dermatol. 2006;126(Supplement):24.
-
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768–3785. - PubMed
-
- Zackheim HS, Amin S, Kashani-Sabet M, McMillan A. Prognosis in cutaneous T-cell lymphoma by skin stage: long-term survival in 489 patients. J Am Acad Dermatol. 1999;40:418–425. - PubMed
-
- Diamandidou E, Colome-Grimmer M, Fayad L, Duvic M, Kurzrock R. Transformation of mycosis fungoides/Sezary syndrome: clinical characteristics and prognosis. Blood. 1998;92:1150–1159. - PubMed
-
- Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857–866. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
