

RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells
- PMID: 17638924
- PMCID: PMC4151289
- DOI: 10.1093/carcin/bgm159
RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells
Abstract
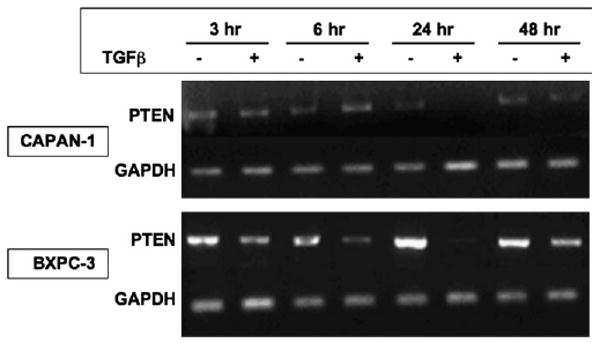
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is rarely mutated in pancreatic cancers, but its regulation by transforming growth factor (TGF)-beta might mediate growth suppression and other oncogenic actions. Here, we examined the role of TGFbeta and the effects of oncogenic K-RAS/ERK upon PTEN expression in the absence of SMAD4. We utilized two SMAD4-null pancreatic cell lines, CAPAN-1 (K-RAS mutant) and BxPc-3 (WT-K-RAS), both of which express TGFbeta surface receptors. Cells were treated with TGFbeta1 and separated into cytosolic/nuclear fractions for western blotting with phospho-SMAD2, SMAD 2, 4 phospho-ATP-dependent tyrosine kinases (Akt), Akt and PTEN antibodies. PTEN mRNA levels were assessed by reverse transcriptase-polymerase chain reaction. The MEK1 inhibitor, PD98059, was used to block the downstream action of oncogenic K-RAS/ERK, as was a dominant-negative (DN) K-RAS construct. TGFbeta increased phospho-SMAD2 in both cytosolic and nuclear fractions. PD98059 treatment further increased phospho-SMAD2 in the nucleus of both pancreatic cell lines, and DN-K-RAS further improved SMAD translocation in K-RAS mutant CAPAN cells. TGFbeta treatment significantly suppressed PTEN protein levels concomitant with activation of Akt by 48 h through transcriptional reduction of PTEN mRNA that was evident by 6 h. TGFbeta-induced PTEN suppression was reversed by PD98059 and DN-K-RAS compared with treatments without TGFbeta. TGFbeta-induced PTEN expression was inversely related to cellular proliferation. Thus, oncogenic K-RAS/ERK in pancreatic adenocarcinoma facilitates TGFbeta-induced transcriptional down-regulation of the tumor suppressor PTEN in a SMAD4-independent manner and could constitute a signaling switch mechanism from growth suppression to growth promotion in pancreatic cancers.
Conflict of interest statement
Figures







Similar articles
-
TGFbeta modulates PTEN expression independently of SMAD signaling for growth proliferation in colon cancer cells.Cancer Biol Ther. 2008 Oct;7(10):1694-9. doi: 10.4161/cbt.7.10.6665. Epub 2008 Oct 22. Cancer Biol Ther. 2008. PMID: 18769113 Free PMC article.
-
TGF-beta downregulates PTEN via activation of NF-kappaB in pancreatic cancer cells.Am J Physiol Gastrointest Liver Physiol. 2010 Feb;298(2):G275-82. doi: 10.1152/ajpgi.00344.2009. Epub 2009 Nov 25. Am J Physiol Gastrointest Liver Physiol. 2010. PMID: 19940030 Free PMC article.
-
TGFbeta1 represses proliferation of pancreatic carcinoma cells which correlates with Smad4-independent inhibition of ERK activation.Oncogene. 2000 Sep 14;19(39):4531-41. doi: 10.1038/sj.onc.1203806. Oncogene. 2000. PMID: 11002426
-
Role of Ras and Mapks in TGFbeta signaling.Cytokine Growth Factor Rev. 2000 Mar-Jun;11(1-2):23-35. doi: 10.1016/s1359-6101(99)00026-x. Cytokine Growth Factor Rev. 2000. PMID: 10708950 Review.
-
Two sides of the story? Smad4 loss in pancreatic cancer versus head-and-neck cancer.FEBS Lett. 2012 Jul 4;586(14):1984-92. doi: 10.1016/j.febslet.2012.01.054. Epub 2012 Feb 3. FEBS Lett. 2012. PMID: 22321641 Free PMC article. Review.
Cited by
-
Genetic status of KRAS modulates the role of Neuropilin-1 in tumorigenesis.Sci Rep. 2017 Oct 10;7(1):12877. doi: 10.1038/s41598-017-12992-2. Sci Rep. 2017. PMID: 29018205 Free PMC article.
-
Clinicopathological significance of SMAD4 loss in pancreatic ductal adenocarcinomas: a systematic review and meta-analysis.Oncotarget. 2017 Mar 7;8(10):16704-16711. doi: 10.18632/oncotarget.14335. Oncotarget. 2017. PMID: 28053288 Free PMC article.
-
TGFβ-Responsive HMOX1 Expression Is Associated with Stemness and Invasion in Glioblastoma Multiforme.Stem Cells. 2016 Sep;34(9):2276-89. doi: 10.1002/stem.2411. Epub 2016 Jul 4. Stem Cells. 2016. PMID: 27354342 Free PMC article.
-
Loss of phosphatase activity in PTEN (phosphatase and tensin homolog deleted on chromosome ten) results in endometrial carcinoma in humans: An in-silico study.Heliyon. 2020 Jan 6;6(1):e03106. doi: 10.1016/j.heliyon.2019.e03106. eCollection 2020 Jan. Heliyon. 2020. PMID: 32042934 Free PMC article.
-
Signaling via Alk5 controls the ontogeny of lung Clara cells.Development. 2010 Mar;137(5):825-33. doi: 10.1242/dev.040535. Development. 2010. PMID: 20147383 Free PMC article.
References
-
- Warshaw AL, et al. Pancreatic carcinoma. N Engl J Med. 1992;326:455–465. - PubMed
-
- Friess H, et al. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105:1846–1856. - PubMed
-
- Derynck R, et al. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. - PubMed
-
- Pasche B. Role of transforming growth factor beta in cancer. J Cell Physiol. 2001;186:153–168. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
