

Establishment and characterization of Fabry disease endothelial cells with an extended lifespan
- PMID: 17644384
- PMCID: PMC2063578
- DOI: 10.1016/j.ymgme.2007.06.003
Establishment and characterization of Fabry disease endothelial cells with an extended lifespan
Abstract
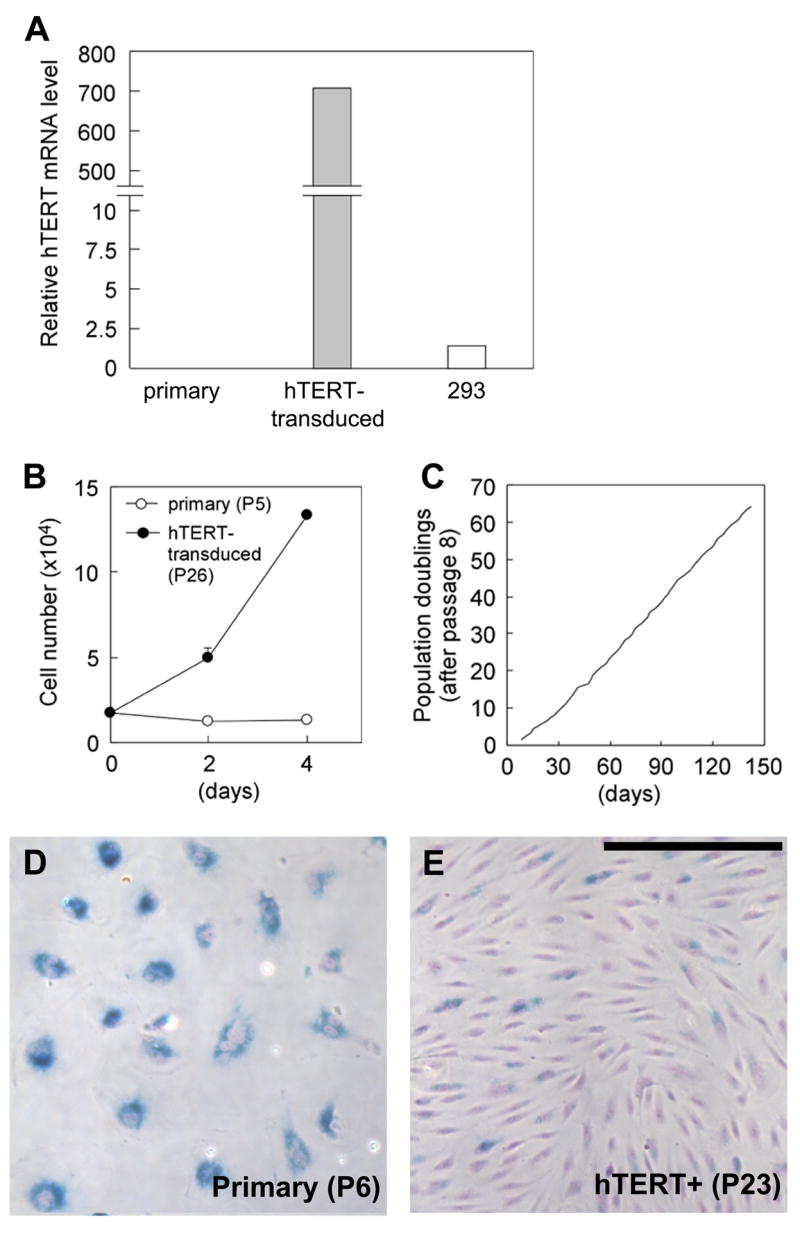
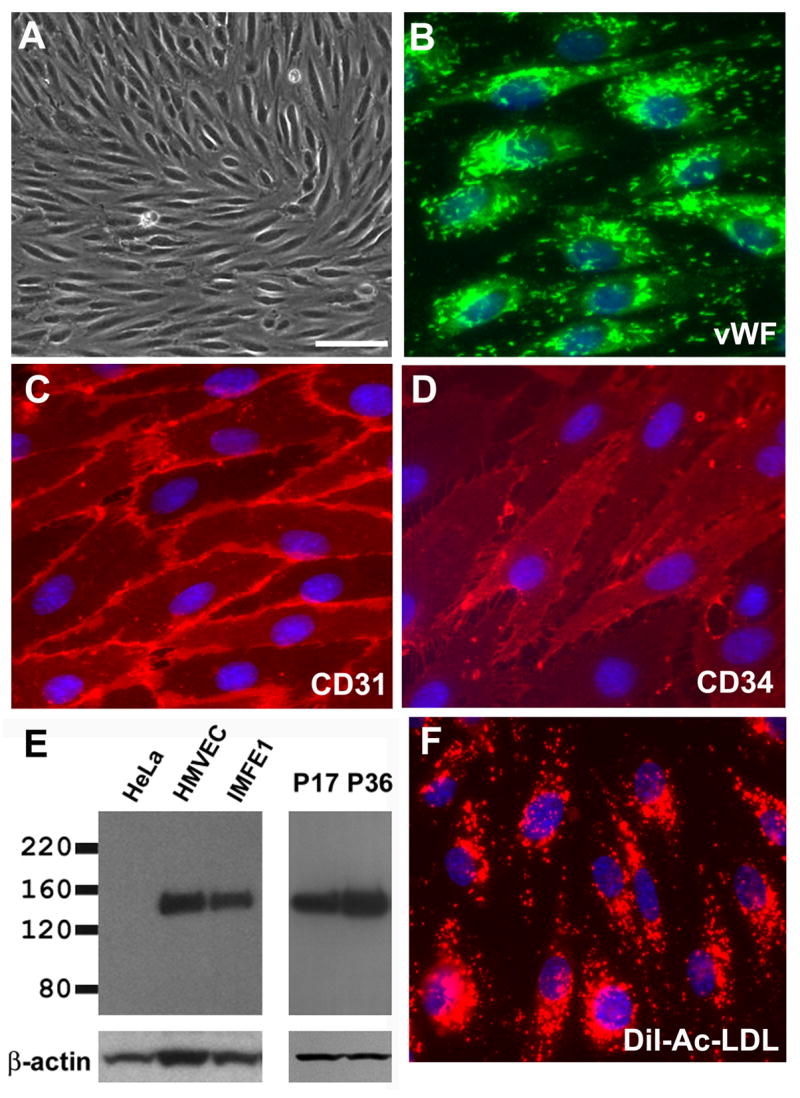
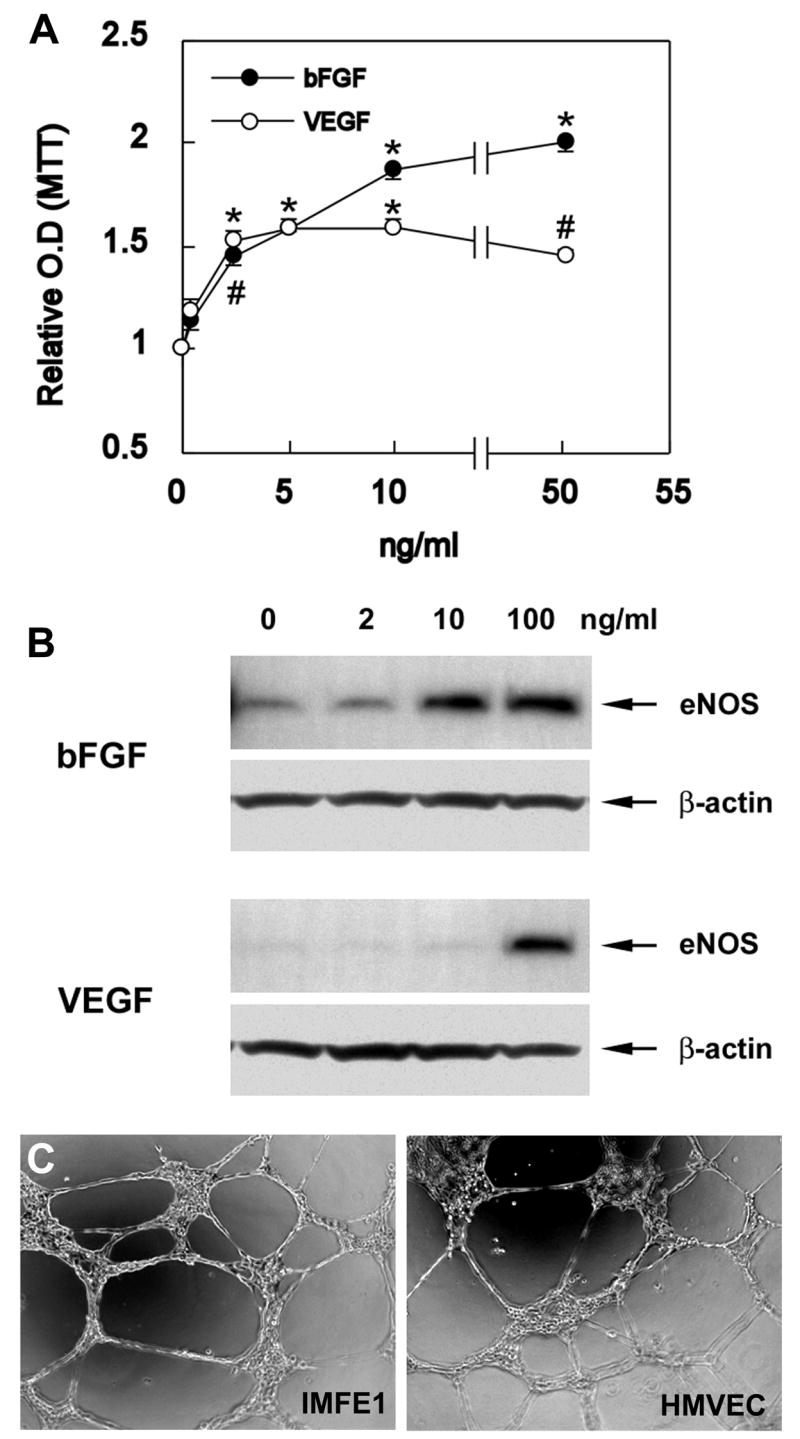
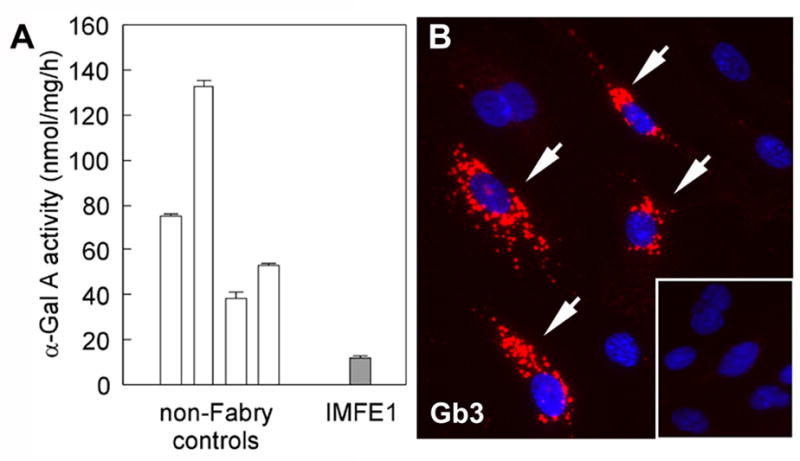
Fabry disease is an inborn error of glycosphingolipid catabolism resulting from a deficiency of lysosomal enzyme alpha-galactosidase A. The major clinical manifestations of the disease, such as stroke, cardiac dysfunction, and renal impairment, are thought to be caused by vasculopathy due to progressive accumulation of globotriaosylceramide in vascular endothelial cells. The pathogenesis of the vasculopathy has not been elucidated. Since in vitro studies using primary endothelial cells are hampered by the limited lifespan of these cells, the availability of cultured endothelial cells with an extended lifespan is critical for the study of the vasculopathy of Fabry disease. We therefore generated an endothelial cell line from a Fabry hemizygote by introduction of human telomerase reverse transcriptase gene. The cell line has markedly extended lifespan compared to parental primary cells. The cells stably express many key markers of endothelial cells such as von Willebrand factor, CD31, CD34, and endothelial nitric oxide synthase (eNOS) and retain functional characteristics such as uptake of acetylated low-density lipoprotein, responsiveness to angiogenic growth factors, up-regulation of eNOS production upon extracellular stimuli, and formation of tube-like structures on Matrigel basement membrane matrix. The cells show significantly reduced activity of alpha-galactosidase A compared with primary endothelial cells from normal individuals and accumulate globotriaosylceramide in lysosomes. This cell line will provide a useful in vitro model of Fabry disease and will facilitate systematic studies to investigate pathogenic mechanisms and explore new therapeutic approaches for Fabry disease.
Figures
References
-
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramide trihexosidase deficiency. N Engl J Med. 1967;276:1163–1167. - PubMed
-
- Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. McGraw-Hill; New York: 2001. pp. 3733–3774.
-
- Moore DF, Kaneski CR, Askari H, Schiffmann R. The cerebral vasculopathy of Fabry disease. J Neurol Sci. 2007 [Epub ahead of print] - PubMed
-
- Heare T, Alp NJ, Priestman DA, Kulkarni AB, Qasba P, Butters TD, Dwek RA, Clarke K, Channon KM, Platt FM. Severe endothelial dysfunction in the aorta of a mouse model of Fabry disease; partial prevention by N-butyldeoxynojirimycin treatment. J Inherit Metab Dis. 2007;30:79–87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
