

Endpoints for clinical trials in young children with cystic fibrosis
- PMID: 17652509
- PMCID: PMC2647606
- DOI: 10.1513/pats.200703-041BR
Endpoints for clinical trials in young children with cystic fibrosis
Abstract
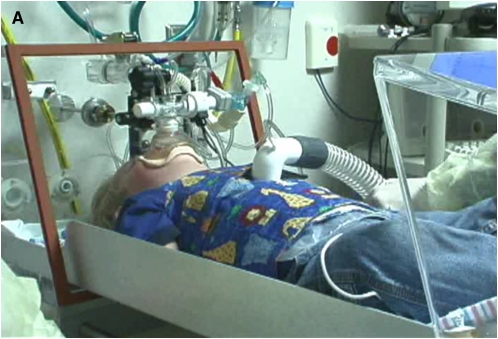
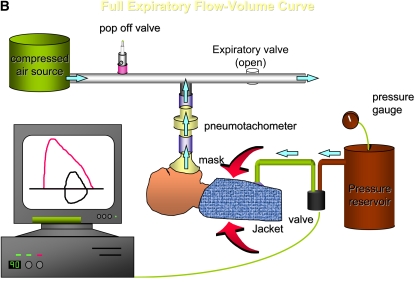
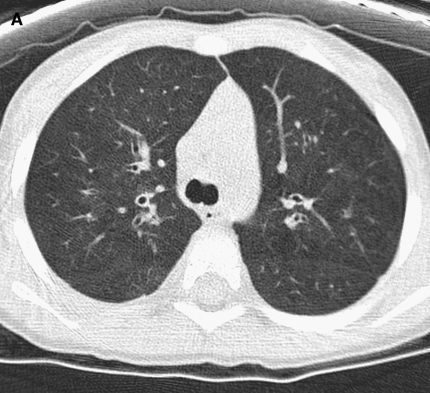
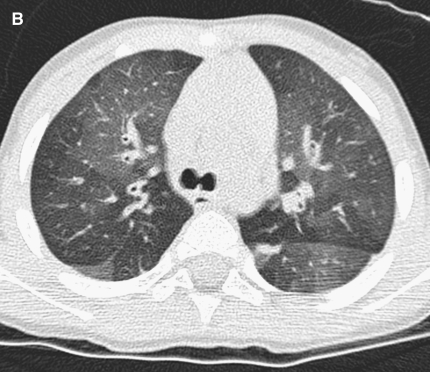
The availability of sensitive, reproducible, and feasible outcome measures for quantifying lung disease in children with cystic fibrosis (CF) younger than 6 years is critical to the conduct of clinical trials in this important population. Historically, identifying and quantifying the presence of lung disease in very young children with CF was hampered by a lack of reproducible measures of lung function or lung pathology. Over the past 10 years, significant progress has led to physiologic, anatomic, and bronchoscopic measures that may serve as endpoints for future intervention trials. These endpoints include infant and preschool lung function testing, computed tomography of the chest, and bronchoalveolar lavage markers of inflammation and infection. Much progress has occurred in standardizing lung function testing, which is essential for multicenter collaboration. Pulmonary exacerbation has the potential to serve as a clinical endpoint; however, there is currently no standardized definition in children with CF younger than 6 years. Further development of these outcomes measures will enable clinical trials in the youngest CF population with the objective of improving long-term prognosis.
Figures
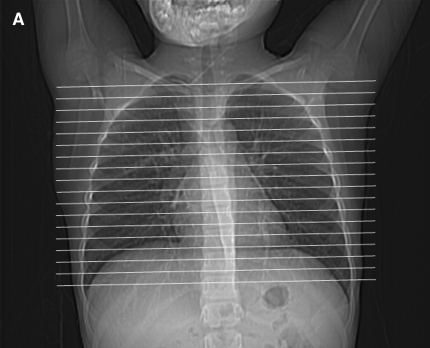
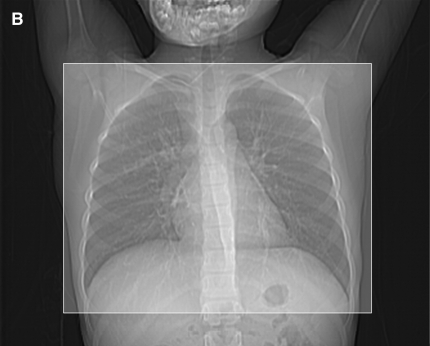
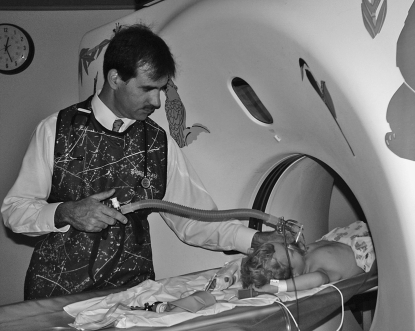
References
-
- Hammer J, Eber E, editors. Paediatric pulmonary function testing. Basel: Karger; 2005.
-
- Gappa M, Ranganathan S, Stocks J. Lung function testing in infants with cystic fibrosis: lessons from the past and future directions. Pediatr Pulmonol 2001;32:228–245. - PubMed
-
- Tepper RS, Montgomery GL, Ackerman V, Eigen H. Longitudinal evaluation of pulmonary function in infants and very young children with cystic fibrosis. Pediatr Pulmonol 1993;16:96–100. - PubMed
-
- Tepper RS, Hiatt P, Eigen H, Scott P, Grosfeld J, Cohen M. Infants with cystic fibrosis: pulmonary function at diagnosis. Pediatr Pulmonol 1988;5:15–18. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous